« Previous
Next »
(261 hits, 1/1)
Showing
10, 25, 50, 100, 500, 1000, all papers per page.
Sort by:
last publication date,
older publication date,
last update date.
- 1. Phys. Rev. 98, 915 (1955) , “Theory of Donor States in Silicon”, W. Kohn, J. M. Luttinger.By using the recently measured effective masses for n-type Si, m1=0.98 m and m2=0.19 m, approximate solutions of the resulting effective mass Schroedinger equation are obtained. The accuracy of the solutions was tested in the limiting cases where... (Read more)
- 2. Solid State Physics 5, 258-319 (1957) , Academic Press, New York (Edited by F. Seitz, D. Turnbull) , “Shallow Impurity States in Silicon and Germanium”, W. KohnI. Introduction (p.258): II. Emprical Properties (p.261): 1. Energy Levels (p.261), a. Ionization Energies, b. Spectra of Excited States, 2. Spin Resonance (p.266), a. Electron Spin Resonance, b. Double Resonance, 3. Static Magnetic Susceptibility (p.271), III. Structure of Donor States (p.271): 4. Conduction Bands of Silicon and Germanium (p.271), a. Silicon, b. Germanium, 5. Effective Mass Theory of Donor States (p.274), a. Single Band Minimum at k=0, b. Several Conduction Band Minima, c. Matrix Elements for Radiative Transitions, 6. Numerical Results and Comparison with Experiments (p.285), a. Energy Levels, b. Wave Functions, 7. Corrections to the Effective Mass Formalism (p.289), a. General Considerations, b. Corrected Wave Functions, c. Comparison with Experiment, IV. Structure of Acceptor States (p.297): 8. Valence Bands of Silicon and Germanium (p.297), a. Silicon, b. Germanium, 9. Effective Mass Equations for Acceptor States (p.300), 10. Approximate Solutions and Comparison with Experiment (p.301) a. Germanium b. Silicon V.Effects of Strains and of Static Electric and Magnetic Fields (p.306): 11. Strains (p.306) a. Donor States, b. Acceptor States, 12. Stark Effect (p.311)
- 3. Sov. Phys. Solid State 23, 2126 (1981) , “Electron spin resonance of exchange-coupled vacancy pairs in hexagonal silicon carbide”, V. S. Va?ner, V. A. ll?in
- 4. J. Appl. Phys. 54, 179-183 (1983) , “The Mechanism of the Enhancement of Divacancy Production by Oxygen During Electron Irradiation of Silicon. II. Computer Modeling”, G. S. Oehrlein, I. Krafcsik, J. L. Lindström, A. E. Jaworowski, and J. W. CorbettNumerical tests of possible models for the oxygen dependence of the divacancy introduction rate in silicon electron irradiated at room temperature were performed on a computer. Only the model in which oxygen traps Si self-interstitials can reproduce all the experimental data. Our modeling results... (Read more)
- 5. Phys. Rev. B 34, 3610-3619 (1986) , “Dipolar interactions between dangling bonds at the (111) Si-SiO2 interface”, K. L. Brower, T. J. HeadleyIn this paper a computational model is developed which allows one to calculate the contribution to the Zeeman linewidth arising from magnetic dipole-dipole interactions between unpaired electrons in the dilute limit, which in our specific application correspond to dangling bonds (Pb... (Read more)
- 6. J. Appl. Phys. 59, 3255-3266 (1986) , “Thermodynamic and Kinetic Considerations on the Equilibrium Shape for Thermally Induced Microdefects in Czochralski Silicon”, W. A. Tiller, S. Hahn, F. A. Ponce.Using thermodynamic and kinetic considerations, we explain the quasiequilibrium, morphological, and structural characteristics of thermally induced oxide precipitates in Czochralski silicon. A model based upon the formation of Frenkel defects at the silicon/silica interface is used to explain the... (Read more)
- 7. Phys. Rev. B 36, 9638-9648 (1987) , “Theory of the Pb center at the <111> Si/SiO2 interface”, A. H. EdwardsWe present a series of semiempirical calculations on threefold-coordinated silicon at the ?111? Si/SiO2 interface. These were performed on finite clusters of atoms with use of hydrogen terminators in an unrestricted Hartree-Fock formalism wherein we include lattice relaxations. We have... (Read more)
- 8. Phys. Rev. B 38, 12752(R) (1988) , “Formation energies, abundances, and the electronic structure of native defects in cubic SiC”, C. Wang, J. Bernholc, R. F. DavisThe relative abundance of native point defects in cubic SiC has been studied via ab initio calculations as a function of composition and the Fermi-level position. For Si-rich cubic SiC, the SiC antisite is the dominant defect in n-type material, while the carbon vacancy, which is a double... (Read more)
- 9. Phys. Rev. B 38, 9674-9685 (1988) , “Hyperfine interactions in cluster models of the Pb defect center”, M. Cook, C. T. WhiteHyperfine interactions in the Pb center (denoted schematically as Si3?Si?) at the Si(111)/SiO2 interface have been studied with use of spin-polarized self-consistent multiple-scattering X? calculations on Si22H21/Si6O18... (Read more)
- 10. Phys. Rev. B 39, 10791-10808 (1989) , “Theory of hydrogen diffusion and reactions in crystalline silicon”, Chris G. Van de Walle, P. J. H. Denteneer, Y. Bar-Yam, and S. T. PantelidesThe behavior of hydrogen in crystalline silicon is examined with state-of-the-art theoretical techniques, based on the pseudopotential-density-functional method in a supercell geometry. Stable sites, migration paths, and barriers for different charge states are explored and displayed in total-energy... (Read more)
- 11. Phys. Rev. B 46, 12335 (1992) , “Microscopic mechanism of atomic diffusion in Si under pressure ”, Osamu Sugino and Atsushi OshiyamaWe have performed the first-principles total-energy calculations on the atomic diffusion of group-V impurities in Si, and have revealed the pressure effect on the activation energy of the diffusion. For the vacancy mechanism, the activation energies for P, As, and Sb decrease with pressure. For the... (Read more)
- 12. Phys. Rev. B 47, 3620-3625 (1993) , “{H,B}, {H,C}, and {H,Si} pairs in silicon and germanium”, Dj. M. Maric, P. F. Meier, S. K. EstreicherThe interactions between interstitial H and substitutional B, C, and Si in crystalline silicon and germanium are studied in molecular clusters at the ab initio Hartree-Fock level with large basis sets. The energetics, electronic structures, and relative stabilities of these pairs are determined. Our... (Read more)
- 13. Phys. Lett. A 213, 89-92 (1996) , “Ground-state zero-field splitting of Mn2+ ions in ZnO and CdSe crystals”, Xiao-Yu KuangZnO and CdSe crystals have similar hexagonal wurtzite structures with a contraction along the c-axis of the crystal, but contrary electronic fine structures for ZnO:Mn2+ (D < 0) and CdSe:Mn2+ (D > 0) have been found in EPR experiments. We demonstrate that the ground-state splitting in ZnO:Mn2+... (Read more)
- 14. phys. stat. sol. (a) 162, 95-151 (1997) , “EPR and ENDOR Investigations of Shallow Impurities in SiC Polytypes”, S. Greulich-WeberInvestigations of nitrogen donors in 6H-, 4H- and 3C-SiC using conventional electron paramagnetic resonance (EPR), electron nuclear double resonance (ENDOR) and optical detection of EPR and ENDOR as well as optical absorption and emission spectroscopy are reviewed and critically discussed. An... (Read more)
- 15. Phys. Rev. B 55, 2188-2194 (1997) , “Dynamic Properties of Interstitial Carbon and Carbon-Carbon Pair Defects in Silicon”, P. Leary, R. Jones, S. ?berg, V. J. B. Torres.Interstitial carbon, Ci, defects in Si exhibit a number of unexplained features. The Ci defect in the neutral charge state gives rise to two almost degenerate vibrational modes at 920 and 931 cm-1 whose 2:1 absorption intensity ratio naturally suggests a trigonal... (Read more)
- 16. Phys. Rev. B 56, 7384 (1997) , “Negatively charged Si vacancy in 4H SiC: A comparison between theory and experiment”, T. Wimbauer, B. K. Meyer, A. Hofstaetter, A. Scharmann, H. OverhofWe use electron paramagnetic resonance and electron nuclear double resonance to identify the negatively charged Si vacancy in neutron-irradiated 4H SiC. The identification is based on resolved ligand hyperfine interactions with carbon and silicon nearest and next nearest neighbors and on the... (Read more)
- 17. phys. stat. sol. (b) 210, 13 (1998) , “Neutral Vacancies in Group-IV Semiconductors”, A. Zywietz, J. Furthm?ller, F. BechstedtAb initio plane-wave-supercell calculations are performed for the neutral monovacancies in silicon, silicon carbide and diamond using ultrasoft non-normconserving Vanderbilt pseudopotentials. We study the structure, the energetics and the single-particle energy spectrum. The local symmetry, the... (Read more)
- 18. phys. stat. sol. (b) 210, 415-427 (1998) , “The Microscopic and Electronic Structure of Shallow Donors in SiC”, S. Greulich-WeberNitrogen donors in 6H-, 4H- and 3C-SiC were investigated using conventional electron paramagnetic resonance (EPR) and electron nuclear double resonance (ENDOR) and the experimental results are discussed. An attempt is presented to interpret the experimentally found large differences in hyperfine... (Read more)
- 19. Phys. Rev. B 57, 6243 (1998) , “Antisites in silicon carbide”, L. Torpo, S. P?ykk?, and R. M. NieminenTen years ago, deep-level-transient-spectroscopy (DLTS) signals, assigned to centers labeled as H1, H2, H3, and E2, have been detected in neutron-irradiated 3C SiC. The H centers were believed to be the primary point defects and the E2 center a secondary defect, which forms after the H centers start... (Read more)
- 20. Phys. Rev. B 58, 9845 (1998) , “Theory of Carbon-Carbon Pairs in Silicon”, R. B. Capaz, A. Dal Pino, Jr., J. D. Joannopoulos.Interstitial-substitutional carbon pairs (CiCs) in silicon display interesting metastable behavior associated with two different structural configurations. In this work, we perform extensive ab initio calculations on this system. Our results show the following. (i) The... (Read more)
- 21. Nature 396, 58-60 (1998) , “Interface structure between silicon and its oxide by first-principles molecular dynamics”, A. Pasquarello, M. S. Hybertsen, R. CarThe requirement for increasingly thin (<50 Å) insulating oxide layers in silicon-based electronic devices highlights the importance of characterizing the Si–SiO2 interface structure at the atomic scale. Such a characterization relies to a large extent on an understanding of the atomic-scale mechanisms that govern the oxidation process. The widely used Deal–Grove model invokes a two-step process in which oxygen first diffuses through the amorphous oxide network before attacking the silicon substrate, resulting in the formation of new oxide at the buried interface1. But it remains unclear how such a process can yield the observed near-perfect interface2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12. Here we use first-principles molecular dynamics13, 14, 15 to generate a model interface structure by simulating the oxidation of three silicon layers. The resulting structure reveals an unexpected excess of silicon atoms at the interface, yet shows no bonding defects. Changes in the bonding network near the interface occur during the simulation via transient exchange events wherein oxygen atoms are momentarily bonded to three silicon atoms — this mechanism enables the interface to evolve without leaving dangling bonds. (Read more)
- 22. phys. stat. sol. (b) 210, 13 (1999) , “Neutral Vacancies in Group-IV Semiconductors”, A. Zywietz, J. Furthmüller, F. BechstedtAb initio plane-wave-supercell calculations are performed for the neutral monovacancies in silicon, silicon carbide and diamond using ultrasoft non-normconserving Vanderbilt pseudopotentials. We study the structure, the energetics and the single-particle energy spectrum. The local symmetry, the... (Read more)
- 23. Phys. Rev. Lett. 83, 372 (1999) , “Hydrogen Electrochemistry and Stress-Induced Leakage Current in Silica”, Peter E. Bl?chl and James H. StathisHydrogen-related defects in oxygen-deficient silica, representing the material of a thermal gate oxide, are analyzed using first-principles calculations. Energetics and charge-state levels of oxygen vacancies, hydrogen, and their complexes in the silica framework are mapped out. The neutral hydrogen... (Read more)
- 24. Phys. Rev. B 59, 15166 (1999) , “Vacancies in SiC: Influence of Jahn-Teller distortions, spin effects, and crystal structure”, A. Zywietz, J. Furthm?ller, and F. BechstedtWe present results of first-principles calculations for the neutral and charged Si and C monovacancies in cubic (3C) and hexagonal (4H) SiC. The calculations are based on the density functional theory in the local-density approximation as well as local spin density approximation. Explicitly a... (Read more)
- 25. Mater. Sci. Eng. B 61-62, 593 (1999) , “Electronic structure of the anti-structure pair in 3C–SiC”, L. Torpo and R. M. NieminenWe have studied the anti-structure pair (adjacent carbon CSi and silicon SiC antisites) in 3C–SiC using the plane–wave pseudopotential method. We report results for the formation energies, the ionization levels and the geometry of the relaxed structures of the defect in all its... (Read more)
- 26. Phys. Rev. Lett. 85, 2324-2327 (2000) , “Fast Diffusion of H and Creation of Dangling Bonds in Hydrogenated Amorphous Silicon Studied by in situ ESR”, U. K. Das, T. Yasuda, and S. YamasakiThe interaction of atomic hydrogen with a-Si:H films was studied by means of in situ ESR during H plasma treatment. H diffuses into the a-Si:H film and creates additional Si dangling bonds (∼1013 cm -2). We observed a high diffusion coefficient (>10-10 cm... (Read more)
- 27. Phys. Rev. Lett. 85, 2773-2776 (2000) , “Dangling Bond Defects at Si-SiO2 Interfaces: Atomic Structure of the Pb1 Center”, A. Stirling, A. Pasquarello, J.-C. Charlier, R. CarUsing a first-principles approach, we characterize dangling bond defects at Si-SiO2 interfaces by calculating hyperfine parameters for several relaxed structures. Interface models, in which defect Si atoms remain close to crystalline sites of the substrate upon relaxation, successfully... (Read more)
- 28. Phys. Rev. B 61, 12602 (2000) , “Phosphorus-related deep donor in SiC”, A. Gali, P. De?k, P. R. Briddon, R. P. Devaty, W. J. ChoykeTwo phosphorus centers in SiC, an isolated P atom substituting for Si (PSi) and a Si-site P-atom adjacent to a carbon vacancy (PSi+VC) are investigated by first principle calculations. It is shown that PSi+VC produces a singly occupied donor... (Read more)
- 29. Phys. Rev. B 61, 12939 (2000) , “Dimer of Substitutional Carbon in Silicon Studied by EPR and ab initio Methods”, J. R. Byberg, B. Bech Nielsen, M. Fanciulli, S. K. Estreicher, P. A. Fedders.An EPR signal observed in carbon-doped float-zone silicon after irradiation with 2-MeV electrons at room temperature has been investigated. It represents a defect with S=1/2, an apparently isotropic g factor (=2.0030), and a complicated hyperfine structure from 29Si nuclei in five shells... (Read more)
- 30. Phys. Rev. B 61, 13655 (2000) , “Intravacancy transition energies in 3C- and 4H-SiC”, A. Zywietz, J. Furthm?ller, F. BechstedtSpin-polarized ab initio calculations are used to determine energy differences between ground and excited states of silicon and carbon vacancies in 3C- and 4H-SiC. The calculated transition energies are compared with recent findings from photoluminescence and magnetic-resonance experiments. (Read more)
- 31. Phys. Rev. B 61, 8393-8403 (2000) , “Dissociation Kinetics of Hydrogen-Passivated Pb Defects at the (111)Si/SiO2 Interface”, A. Stesmans.An electron-spin-resonance study has been carried out, both isothermally and isochronically, of the recovery under vacuum annealing from the hydrogen passivated state (symbolized as HPb) of paramagnetic Pb centers (Si3?Si?) at the (111)Si/SiO2... (Read more)
- 32. Phys. Rev. B 62, 12923-12926 (2000) , “Electron paramagnetic resonance of Cu(d9) in GaN”, C. Bozdog, K. H. Chow, G. D. Watkins, H. Sunakawa, N. Kuroda, A. UsuiElectron paramagnetic resonance of Cu2+(d9) has been detected optically in the visible and near-infrared luminescence of wurtzite GaN. Its effective S=1/2 spin Hamiltonian parameters are g‖=±0.20(5), g⊥=+1.549(1), and... (Read more)
- 33. Phys. Rev. B 62, 6854 (2000) , “Spin state of vacancies: From magnetic Jahn-Teller distortions to multiplets”, A. Zywietz, J. Furthm?ller, and F. BechstedtThe spin configuration of isoelectronic vacancies surrounded by first-row atoms is studied within density-functional theory (DFT) using the local spin density approximation. Allowing for a symmetry break in the electronic system, a mixed-spin state is found to be lowest in energy. It is accompanied... (Read more)
- 34. J. Phys.: Condens. Matter 12, 10029-10037 (2000) , “New physics ofthe 30° partial dislocation in silicon revealed through ab initio calculation”, G. Cs?nyi, T. D. Engeness, S. Ismail-Beigi, T. A. Arias.On the basis of ab initio calculation, we propose a new structure for the fundamental excitation of the reconstructed 30° partial dislocation in silicon. This soliton has a rare structure involving a fivefold-coordinated atom near the dislocation core. The unique electronic structure... (Read more)
- 35. J. Phys.: Condens. Matter 12, 3369 (2000) , “Local electronic structure around vacancies and vacancy-antisite complexes in ?-SiC”, G. Cubiotti, Yu. Kucherenko, A. Yaresko, A. Perlov, V. AntonovThe local electronic structure around vacancies and vacancy-antisite complexes in cubic SiC has been calculated by means of the LMTO (linear muffin-tin orbital) method and the supercell approach. In order to improve the description of the electronic structure near the energy gap, the... (Read more)
- 36. Physica B 308-310, 621 (2001) , “Positively charged carbon vacancy in 6H–SiC: EPR study”, V. Ya. Bratus, I. N. Makeeva, S. M. Okulov, T. L. Petrenko, T. T. Petrenko and H. J. von BardelebenThe low-temperature X-band EPR study of Ky1 and Ky2 centers assigned to positively charged carbon vacancy (VC+) in two quasicubic sites of 6H–SiC crystal is presented. The CS symmetry, spin S=1/2 and close coincidence of the g-tensor components have been revealed. The principal values of... (Read more)
- 37. Physica B 308-310, 637 (2001) , “Calculation of hyperfine parameters of positively charged carbon vacancy in SiC”, T. T. Petrenko, T. L. Petrenko, V. Ya. Bratus and J. L. MongeTheoretical simulation of hyperfine parameters for the nearest and next-nearest neighbor atoms of VC+ in SiC has been performed for the cubic and hexagonal clusters. The gradient-corrected all-electron DFT calculations with Becke's three-parameter functional have been performed by the use of split... (Read more)
- 38. Phys. Rev. Lett. 86, 4560-4563 (2001) , “Structure and Generation Mechanism of the Peroxy-Radiacal Defect in Amorphous Silica”, T. Uchino, M. Takahashi, and T. YokoWe provide a new model of the peroxy-radical defect in amorphous silica on the basis of quantum-chemical calculations applied to clusters of atoms to model the defect. In this model, the 29Si hyperfine splittings of the peroxy radical arise from a single silicon, in agreement with the... (Read more)
- 39. Phys. Rev. Lett. 86, 5522-5525 (2001) , “E' Centers in Amorphous SiO2 Revisited: A New Look at an Old Problem”, T. Uchino, M. Takahashi, T. YokoWe present theoretical evidence that the paramagnetic E? defect centers in amorphous silicon dioxide ( a-SiO2) do not have the same microscopic structures as those well-defined in the corresponding crystalline counterparts such as ?-quartz. We then present alternative models of... (Read more)
- 40. Phys. Rev. Lett. 87, 165506 (2001) , “Defect Generation by Hydrogen at the Si-SiO2 Interfaces”, S. N. Rashkeev, D. M. Fleetwood, R. D. Schrimpf, and S. T. PantelidesHydrogen is known to passivate Si dangling bonds at the Si-SiO 2 interface, but the subsequent arrival of H + at the interface causes depassivation of Si-H bonds. Here we report first-principles density functional calculations, showing that, contrary to conventional... (Read more)
- 41. Phys. Rev. Lett. 87, 235501 (2001) , “Thermal Double Donors and Quantum Dots”, J. Coutinho, R. Jones, L. I. Murin, V. P. Markevich, J. L. Lindstr?m, S. ?berg, P. R. Briddon.Combined local mode spectroscopy and ab initio modeling are used to demonstrate for the first time that oxygen atoms in thermal double donors (TDD) in Si are in close proximity. The observed vibrational modes in 16O, 18O, and mixed isotopic samples are consistent with a model... (Read more)
- 42. Phys. Rev. B 63, 165204 (2001) , “g values of effective mass donors in AlxGa1-xN alloys”, M. W. Bayerl, M. S. Brandt, T. Graf, O. Ambacher, J. A. Majewski, M. Stutzmann, D. J. As, K. LischkaElectron spin resonance experiments were performed on Si-doped wurtzite and zinc-blende GaN and Si-doped wurtzite AlxGa1-xN alloys with x=0.15, 0.32, 0.52, 0.75, and 1. For zinc-blende GaN, an isotropic g factor of 1.9475 is found. The g tensors of the silicon effective mass... (Read more)
- 43. Phys. Rev. B 63, 245202 (2001) , “Ab initio density-functional supercell calculations of hydrogen defects in cubic SiC”, B. Aradi, A. Gali, P. De?k, J. E. Lowther, N. T. Son, E. Janz?n, W. J. ChoykeBased on ab initio density-functional calculations in supercells of 3C-SiC, the stable configurations of hydrogen and dihydrogen defects have been established. The calculated formation energies are used to give semiquantitative estimates for the concentration of hydrogen in SiC after chemical vapor... (Read more)
- 44. Phys. Rev. B 64, 245208 (2001) , “Ab initio and empirical-potential studies of defect properties in 3C-SiC”, Fei Gao, Eric J. Bylaska, William J. Weber, L. Ren? CorralesDensity functional theory (DFT) is used to study the formation and properties of native defects in 3C-SiC. Extensive calculations have been carried out to determine the formation of point defects and the stability of self-interstitials. Although there is good agreement in the formation of vacancies... (Read more)
- 45. Phys. Rev. B 64, 245212 (2001) , “Structure of the silicon vacancy in 6H-SiC after annealing identified as the carbon vacancy-carbon antisite pair”, Th. Lingner, S. Greulich-Weber, J.-M. Spaeth, U. Gerstmann, E. Rauls, Z. Hajnal, Th. Frauenheim, H. OverhofWe investigated radiation-induced defects in neutron-irradiated and subsequently annealed 6H-silicon carbide (SiC) with electron paramagnetic resonance (EPR), the magnetic circular dichroism of the absorption (MCDA), and MCDA-detected EPR (MCDA-EPR). In samples annealed beyond the annealing... (Read more)
- 46. J. Phys.: Condens. Matter 13, 6203-6231 (2001) , “Comprehensive ab initio study of properties of monovacancies and antisites in 4H-SiC”, L. Torpo, M. Marlo, T. E. M. Staab, R. M. NieminenWe present results of ab initio calculations for the electronic and atomic structures of monovacancies and antisite defects in 4H-SiC in all possible charge states. The calculations make use of a plane-wave pseudopotential method based on density-functional theory and the local spin-density... (Read more)
- 47. J. Phys.: Condens. Matter 13, L1-L7 (2001) , “Identification of the Tetra-Interstitial in Silicon”, B. J. Coomer, J. P. Goss, R. Jones, S. ?berg, P. R. Briddon.First-principles computational methods are employed to investigate the structural, vibrational, optical and electronic properties of the self-interstitial aggregate, I4 in silicon. We find the defect to be electrically active and identify it with the B3 EPR centre. We... (Read more)
- 48. J. Appl. Phys. 89, 5997-6001 (2001) , “Oxygen Recoil Implant from SiO2 Layers into Single-Crystalline Silicon”, G. Wang, Y. Chen, D. Li, S. Oak, G. Srivastav, S. Banerjee, A. Tasch, P. Merrill, R. Bleiler.It is important to understand the distribution of recoil-implanted atoms and the impact on device performance when ion implantation is performed at a high dose through surface materials into single crystalline silicon. For example, in ultralarge scale integration impurity ions are often implanted... (Read more)
- 49. Appl. Phys. Lett. 78, 1571 (2001) , “Hydrogen Passivation and Activation of Oxygen Complexes in Silicon”, S. N. Rashkeev, M. Di Ventra, and S. T. PantelidesWe report first-principles calculations in terms of which we describe the role of hydrogen in passivating or activating oxygen complexes in Si. In particular we find that attaching H to a pre-existing oxygen cluster can change the electric activity of the cluster. Furthermore, the addition of a... (Read more)
- 50. Phys. Rev. Lett. 88, 205502 (2002) , “Metastability of Amorphous Silicon from Silicon Network Rebonding”, R. Biswas, B. C. Pan, and Y. Y. YeWe propose a network rebonding model for light-induced metastability in amorphous silicon, involving bonding rearrangements of silicon and hydrogen atoms. Nonradiative recombination breaks weak silicon bonds and generates dangling bond?floating bond pairs, with very low activation energies. The... (Read more)
- 51. Phys. Rev. B 65, 085202 (2002) , “Divacancy in 3C- and 4H-SiC: An extremely stable defect”, L. Torpo, T. E. M. Staab, R. M. NieminenUsing first-principles calculations for divacancy defects in 3C- and 4H-SiC, we determine their formation energies and stability, their ionization levels, and relaxed geometries (symmetry point groups) for neutral as well as for charged states. For 4H-SiC all four possible nearest-neighbor divacancy... (Read more)
- 52. Phys. Rev. B 65, 184108 (2002) , “Alphabet luminescence lines in 4H-SiC”, T. A. G. Eberlein, C. J. Fall, R. Jones, P. R. Briddon, S. ?bergFirst-principles density functional calculations are used to investigate antisite pairs in 4H-SiC. We show that they are likely to be formed in close proximity under ionizing conditions, and they possess a donor level and thermal stability consistent with the series of 40 photoluminescent lines... (Read more)
- 53. Phys. Rev. B 66, 024106 (2002) , “Cascade overlap and amorphization in 3C-SiC: Defect accumulation, topological features, and disordering”, F. Gao and W. J. WeberMolecular dynamics (MD) simulations with a modified Tersoff potential have been used to investigate cascade overlap, damage accumulation, and amorphization processes in 3C-SiC over dose levels comparable to experimental conditions. A large number of 10 keV displacement cascades were randomly... (Read more)
- 54. Phys. Rev. B 66, 161202(R) (2002) , “Phosphorus and sulphur doping of diamond”, L. G. Wang and Alex ZungerPrevious calculations on n-type doping of diamond by P and S predicted that S has a shallower level and a higher solubility than P. Our first-principles calculations show that the opposite is true: Phosphorus impurity in diamond gives rise to a shallower donor level, and has a higher bulk solid... (Read more)
- 55. J. Appl. Phys. 91, 8919-8941 (2002) , “Transient Enhanced Diffusion of Boron in Si”, S. C. Jain, W. Schoenmaker, R. Lindsay, P. A. Stolk, S. Decoutere, M. Willander, H. E. Maes.On annealing a boron implanted Si sample at ~800 °C, boron in the tail of the implanted profile diffuses very fast, faster than the normal thermal diffusion by a factor 100 or more. After annealing for a sufficiently long time, the enhanced diffusion saturates. The enhanced diffusion is... (Read more)
- 56. J. Appl. Phys. 92, 889-894 (2002) , “Ramification of micropipes in SiC crystals”, M. Yu. Gutkin, A. G. Sheinerman, T. S. Argunova, J. H. Je, H. S. Kang, Y. Hwu, W.-L. TsaiThe ramification of micropipes is observed using scanning electron microscopy, optical microscopy, and synchrotron x-ray radiography. The conditions for the ramification of dislocated micropipes are determined theoretically within a model when the angles between dislocation lines are small. It is... (Read more)
- 57. Appl. Phys. Lett. 81, 1818 (2002) , “Interface structures generated by negative-bias temperature instability in Si/SiO2 and Si/SiOxNy interfaces”, J. Ushio, T. Maruizumi, K. Kushida-AbdelghafarWe used a density functional method to investigate the mechanism of negative-bias temperature instability (NBTI) and resultant structural changes of Si/SiO2 and Si/SiOxNy interfaces. The reaction energies for the water- and hydrogen-originated... (Read more)
- 58. Appl. Phys. Lett. 81, 1839 (2002) , “Dual behavior of H+ at Si–SiO2 interfaces: Mobility versus trapping”, S. N. Rashkeev, D. M. Fleetwood, R. D. Schrimpf, S. T. PantelidesWe report first-principles calculations showing that protons in the vicinity of a Si–SiO2 interface can behave in two different ways. At an abrupt interface without suboxide bonds (Si–Si bonds at the oxide side of the interface) H+ does not become trapped but migrates... (Read more)
- 59. Physica B 340-342, 15-24 (2003) , “Defects in SiC”, E. Janz?n, I. G. Ivanov, N. T. Son, B. Magnusson, Z. Zolnai, A. Henry, J. P. Bergman, L. Storasta, F. CarlssonRecent results from studies of shallow donors, pseudodonors, and deep level defects in SiC are presented. The selection rules for transitions between the electronic levels of shallow donors in 4H–SiC in the dipole approximation are derived and the ionization energy for the N donor at... (Read more)
- 60. Phys. Rev. Lett. 90, 225502 (2003) , “Z1/Z2 Defects in 4H–SiC”, T. A. G. Eberlein, R. Jones, P. R. BriddonFirst-principles calculations are carried out on models for the Z1/Z2 defects in 4H–SiC which are found in as-grown and irradiated n-type material. We show that an interstitial-nitrogen–interstitial-carbon defect is exceptionally thermally stable,... (Read more)
- 61. Phys. Rev. Lett. 91, 136101 (2003) , “Ab initio Simulations of Homoepitaxial SiC Growth”, M. C. Righi, C. A. Pignedoli, R. Di Felice, C. M. Bertoni, A. CatellaniWe present first-principle calculations on the initial stages of SiC homoepitaxial growth on the –SiC(111)–(×) surface. We show that the nonstoichiometric reconstruction plays a relevant role in favoring the attainment of high-quality films. The motivation is twofold: On one hand, we... (Read more)
- 62. Phys. Rev. B 67, 155203 (2003) , “Correlation between the antisite pair and the DI center in SiC”, A. Gali, P. Deák, E. Rauls, N. T. Son, I. G. Ivanov, F. H. C. Carlsson, E. Janzén, and W. J. ChoykeThe DI low temperature photoluminescence center is a well-known defect stable up to 1700 °C annealing in SiC, still its structure is not yet known. Combining experimental and theoretical studies, in this paper we will show that the properties of an antisite pair can reproduce... (Read more)
- 63. Phys. Rev. B 67, 165212 (2003) , “Optical selection rules for shallow donors in 4H-SiC and ionization energy of the nitrogen donor at the hexagonal site”, I. G. Ivanov, B. Magnusson, and E. JanzénThe selection rules for transitions between the electronic levels of shallow donors in 4H-SiC in the dipole approximation are derived. The ionization energy of the shallow nitrogen donor (at hexagonal site) is determined to be 61.4±0.5 meV by analyzing the photothermal ionization and... (Read more)
- 64. Phys. Rev. B 67, 193102 (2003) , “Signature of intrinsic defects in SiC: Ab initio calculations of hyperfine tensors”, Michel Bockstedte, Matthias Heid, and Oleg PankratovTo reveal the microscopic origin of the spin-resonance centers in 3C- and 4H-SiC, we perform first-principles calculations of the hyperfine tensors for vacancy-related defects and interstitials. The calculations for the silicon vacancy corroborates the earlier experimental identification. The... (Read more)
- 65. Phys. Rev. B 67, 205202 (2003) , “Formation and annealing of nitrogen-related complexes in SiC”, U. Gerstmann, E. Rauls, Th. Frauenheim, and H. OverhofWe propose a mechanism for the annealing of vacancy-related defects in SiC, based on ab initio total energy calculations. Our mechanism is based on the formation and migration of carbon and nitrogen split interstitials resulting in CSi(NC)n or... (Read more)
- 66. Phys. Rev. B 68, 085202 (2003) , “Physics and chemistry of hydrogen in the vacancies of semiconductors”, Bernadett Szûcs, Adam Gali, Zoltán Hajnal, Peter Deák, and Chris G. Van de WalleHydrogen is well known to cause electrical passivation of lattice vacancies in semiconductors. This effect follows from the chemical passivation of the dangling bonds. Recently it was found that H in the carbon vacancy of SiC forms a three-center bond with two silicon neighbors in the vacancy, and... (Read more)
- 67. Phys. Rev. B 68, 125201 (2003) , “Aggregation of carbon interstitials in silicon carbide: A theoretical study”, A. Gali, P. Deák, P. Ordejón, N. T. Son, E. Janzén, and W. J. ChoykeAb initio supercell calculations have been carried out to investigate clusters of carbon interstitials in 3C- and 4H-SiC. Based on the calculated formation energies, the complex formation of carbon interstitials or their aggregation to carbon antisites is energetically favored... (Read more)
- 68. Phys. Rev. B 68, 125204 (2003) , “Magnetic properties of substitutional 3d transition metal impurities in silicon carbide”, M. S. Miao and Walter R. L. LambrechtUsing the linearized muffin-tin orbital (LMTO) method within both the atomic sphere approximation and full potential (FP) implementations and within the local spin-density-functional method and a supercell approach, we study the magnetic properties of cubic (3C) silicon carbide (SiC) doped by... (Read more)
- 69. Phys. Rev. B 68, 155208 (2003) , “Theoretical study of vacancy diffusion and vacancy-assisted clustering of antisites in SiC”, E. Rauls, Th. Frauenheim, A. Gali, P. De?kUsing the self-consistent-charge density-functional-based tight-binding (SCC-DFTB) method, we have investigated the migration of vacancies at high temperatures, taking into account the entropy contribution to the Gibbs free energy. We have found that the energy barrier for sublattice migration of... (Read more)
- 70. Phys. Rev. B 68, 205201 (2003) , “Ab initio study of the migration of intrinsic defects in 3C-SiC”, Michel Bockstedte, Alexander Mattausch, and Oleg PankratovThe diffusion of intrinsic defects in 3C-SiC is studied using an ab initio method based on density functional theory. The vacancies are shown to migrate on their own sublattice. The carbon split-interstitials and the two relevant silicon interstitials, namely the tetrahedrally... (Read more)
- 71. J. Appl. Phys. 94, 1 (2003) , “Negative bias temperature instability: Road to cross in deep submicron silicon semiconductor manufacturing”,We present an overview of negative bias temperature instability (NBTI) commonly observed in p-channel metal–oxide–semiconductor field-effect transistors when stressed with negative gate voltages at elevated temperatures. We discuss the results of such stress on device and circuit... (Read more)
- 72. Phys. Rev. Lett. 93, 086102 (2004) , “Reaction of the Oxygen Molecule at the Si(100)–SiO2 Interface During Silicon Oxidation”, Angelo Bongiorno and Alfredo PasquarelloUsing constrained ab initio molecular dynamics, we investigate the reaction of the O2 molecule at the Si(100)–SiO2 interface during Si oxidation. The reaction proceeds sequentially through the incorporation of the O2 molecule in a Si-Si bond and the... (Read more)
- 73. Phys. Rev. Lett. 93, 245901 (2004) , “Ab Initio Calculations to Model Anomalous Fluorine Behavior”, Milan Diebel, Scott T. Dunhammplanted fluorine is observed to behave unusually in silicon, manifesting apparent uphill diffusion and reducing diffusion and enhancing activation of boron. In order to investigate fluorine behavior, we calculate the energy of fluorine defect structures in the framework of density functional theory. In addition to identifying the ground-state configuration and diffusion migration barrier of a single fluorine atom in silicon, a set of energetically favorable fluorine defect structures were found (FnVm). The decoration of vacancies and dangling silicon bonds by fluorine suggests that fluorine accumulates in vacancy-rich regions, which explains the fluorine redistribution behavior reported experimentally. (Read more)
- 74. Phys. Rev. B 69, 045322 (2004) , “Carbon antisite clusters in SiC: A possible pathway to the DII center”, Alexander Mattausch, Michel Bockstedte, and Oleg PankratovThe photoluminescence center DII is a persistent intrinsic defect which is common in all SiC polytypes. Its fingerprints are the characteristic phonon replicas in luminescence spectra. We perform ab initio calculations of vibrational spectra for various defect complexes and... (Read more)
- 75. Phys. Rev. B 69, 125203 (2004) , “First-principles studies of the diffusion of B impurities and vacancies in SiC”, R. Rurali, E. Hernández, P. Godignon, J. Rebollo, and P. OrdejónIn this paper we analyze, by means of first-principles electronic structure calculations, the structural, energetic, and diffusive properties of B impurities in SiC as well as of vacancies. We focus our study on (i) determining the equilibrium structures of the impurity in the lattice by means of... (Read more)
- 76. Phys. Rev. B 69, 233202 (2004) , “Diffusion of hydrogen in perfect, p-type doped, and radiation-damaged 4H-SiC”, B. Aradi, P. De?k, A. Gali, N. T. Son, E. Janz?nThe diffusion of interstitial atomic hydrogen in 4H-SiC was investigated theoretically, using the local density approximation of density functional theory. We have found that the diffusion barrier in the perfect crystal is 0.6 eV. Comparing this value with the calculated zero point vibration... (Read more)
- 77. Phys. Rev. B 69, 235202 (2004) , “Ab initio study of the annealing of vacancies and interstitials in cubic SiC: Vacancy-interstitial recombination and aggregation of carbon interstitials”, Michel Bockstedte, Alexander Mattausch, and Oleg PankratovThe annealing kinetics of mobile intrinsic defects in cubic SiC is investigated by an ab initio method based on density-functional theory. The interstitial-vacancy recombination, the diffusion of vacancies, and interstitials to defect sinks (e.g., surfaces or dislocations) as well as the... (Read more)
- 78. Phys. Rev. B 69, 245205 (2004) , “Atomistic study of intrinsic defect migration in 3C-SiC”, Fei Gao, William J. Weber, M. Posselt, and V. BelkoAtomic-scale computer simulations, both molecular dynamics (MD) and the nudged-elastic band methods, have been applied to investigate long-range migration of point defects in cubic SiC (3C-SiC) over the temperature range from 0.36Tm to 0.95Tm (melting... (Read more)
- 79. Phys. Rev. B 70, 085202 (2004) , “Reassignment of phosphorus-related donors in SiC”, E. Rauls, M. V. B. Pinheiro, S. Greulich-Weber, and U. GerstmannCombining efficient density-functional based tight-binding molecular dynamics with ab initio calculations, we show that despite higher formation energies the incorporation of phosphorus at the carbon sublattice is favored by kinetic effects during the annealing processes. Based on the... (Read more)
- 80. Phys. Rev. B 70, 115203 (2004) , “Different roles of carbon and silicon interstitials in the interstitial-mediated boron diffusion in SiC”, Michel Bockstedte, Alexander Mattausch, and Oleg PankratovThe interstitial and vacancy mediated boron diffusion in silicon carbide is investigated with an ab initio method. The boron interstitials in p-type and n-type materials are found to be far more mobile than the boron-vacancy complexes. A kick-out mechanism and an interstitialcy... (Read more)
- 81. Phys. Rev. B 70, 195344 (2004) , “Ab initio study of structural and electronic properties of planar defects in Si and SiC”, C. Raffy, J. Furthmüller, J.-M. Wagner, and F. BechstedtWe present ab initio calculations for internal interfaces in Si and SiC. Density-functional calculations within the local-density approximation and the pseudopotential-plane-wave approach are performed to understand the effect of such two-dimensional defects on the electronic properties. We... (Read more)
- 82. Phys. Rev. B 70, 201204(R) (2004) , “Annealing of vacancy-related defects in semi-insulating SiC”, U. Gerstmann, E. Rauls, and H. OverhofThe annealing of P6/P7 centers (VCCSi pairs) in the presence of carbon vacancies in high concentrations typical for semi-insulating (SI) silicon carbide (SiC) is studied theoretically. The calculated hyperfine parameters support the suggestion of a negatively... (Read more)
- 83. Phys. Rev. B 70, 205207 (2004) , “Hydrogen-related photoluminescent centers in SiC”, D. Prezzi, T. A. G. Eberlein, R. Jones, B. Hourahine, P. R. Briddon, S. ?bergLocal density functional calculations are used to investigate models of the center responsible for a prominent set of luminescent lines with zero-phonon lines around 3.15 eV in hydrogen rich 4H-SiC and previously attributed to VSi-H. We find that the electronic structure of... (Read more)
- 84. Phys. Rev. B 70, 235211 (2004) , “Structure and vibrational spectra of carbon clusters in SiC”, Alexander Mattausch, Michel Bockstedte, and Oleg PankratovThe electronic, structural, and vibrational properties of small carbon interstitial and antisite clusters are investigated by ab initio methods in 3C- and 4H-SiC. The defects possess sizable dissociation energies and may be formed via condensation of carbon interstitials, e.g.,... (Read more)
- 85. Phys. Rev. B 70, 235212 (2004) , “EPR and theoretical studies of positively charged carbon vacancy in 4H-SiC”, T. Umeda, J. Isoya, N. Morishita, T. Ohshima, T. Kamiya, A. Gali, P. De?k, N. T. Son, E. Janz?nThe carbon vacancy is a dominant defect in 4H-SiC, and the "EI5" electron-paramagnetic-resonance (EPR) spectrum originates from positively charged carbon vacancies (VC+) at quasicubic sites. The observed state for EI5, however, has been attributed to a... (Read more)
- 86. Phys. Rev. B 70, 245204 (2004) , “Silicon vacancy annealing and DI luminescence in 6H-SiC”, M. V. B. Pinheiro, E. Rauls, U. Gerstmann, S. Greulich-Weber, H. Overhof, and J.-M. SpaethCombining electron paramagnetic resonance measurements with ab initio calculations, we identify the VCCSi(SiCCSi) complex as a second annealing product of the silicon vacancy via an analysis of resolved carbon hyperfine interactions and of... (Read more)
- 87. Phys .Rev. Lett. 92, 178302 (2004) , “Nonvolatile Memory with Multilevel Switching: A Basic Model”, M. J. Rozenberg, I. H. Inoue, and M. J. SánchezThere is a current upsurge in research on nonvolatile two-terminal resistance random access memory (RRAM) for next generation electronic applications. The RRAM is composed of a simple sandwich of a semiconductor with two metal electrodes. We introduce here an initial model for RRAM with the... (Read more)
- 88. Phys. Rev. B 71, 035213 (2005) , “Possibility for the electrical activation of the carbon antisite by hydrogen in SiC”, A. Gali, P. Deák, N. T. Son, and E. JanzénCalculations predict the carbon antisite to be the most abundant intrinsic defect in silicon carbide in a wide range of doping. The isolated carbon antisite is, however, optically and electronically inactive, therefore, difficult to observe by usual experimental techniques. However, CSi... (Read more)
- 89. Phys. Rev. B 71, 125202 (2005) , “Positively charged carbon vacancy in three inequivalent lattice sites of 6H-SiC: Combined EPR and density functional theory study”, V. Ya. Bratus', T. T. Petrenko, S. M. Okulov, and T. L. PetrenkoThe Ky1, Ky2, and Ky3 centers are the dominant defects produced in the electron-irradiated p-type 6H-SiC crystals. The electron paramagnetic resonance study of these defects has been performed in the temperature range of 4.2–300 K at... (Read more)
- 90. Phys. Rev. B 71, 193202 (2005) , “EPR and theoretical studies of negatively charged carbon vacancy in 4H-SiC”, T. Umeda, Y. Ishitsuka, J. Isoya, N. T. Son, E. Janz?n, N. Morishita, T. Ohshima, H. Itoh, A. GaliCarbon vacancies (VC) are typical intrinsic defects in silicon carbides (SiC) and so far have been observed only in the form of positively charged states in p-type or semi-insulating SiC. Here, we present electron-paramagnetic-resonance (EPR) and photoinduced EPR (photo-EPR)... (Read more)
- 91. Phys. Rev. B 71, 193204 (2005) , “Angular correlation of annihilation radiation associated with vacancy defects in electron-irradiated 6H-SiC”, A. Kawasuso, T. Chiba, T. HiguchiElectron-positron momentum distributions associated with vacancy defects in 6H-SiC after irradiation with 2-MeV electrons and annealing at 1000 °C have been studied using angular correlation of annihilation radiation measurements. It was confirmed that the above vacancy defects have... (Read more)
- 92. Phys. Rev. B 71, 245203 (2005) , “Electrical Activity of Er and Er-O Centers in Silicon”, D. Prezzi, T. A. G. Eberlein, R. Jones, J. S. Filhol, J. Coutindo, M. J. Shaw, P. R. Briddon.Spin-polarized density functional calculations are carried out on Er and Er-oxygen defects in crystalline Si. We find that the interstitial site is favored but the diffusion barrier of Eri is only 1.9 eV, and inevitably Eri forms complexes with impurities and... (Read more)
- 93. Phys. Rev. B 72, 045219 (2005) , “Fluorine in Si: Native-defect complexes and the supression of impurity diffusion”, Giorgia M. Lopez, Vincenzo Fiorentini, Giuliana Impellizzeri, Salvatore Mirabella, Enrico NapolitaniThe transient enhanced diffusion of acceptor impurities severely affects the realization of ultrahigh doping regions in miniaturized Si-based devices. Fluorine codoping has been found to suppress this transient diffusion, but the mechanism underlying this effect is not understood. It has been proposed that fluorine-impurity or fluorine–native-defect interactions may be responsible. Here we clarify this mechanism combining first-principles theoretical studies of fluorine in Si and purposely designed experiments on Si structures containing boron and fluorine. The central interaction mechanism is the preferential binding of fluorine to Si-vacancy dangling bonds and the consequent formation of vacancy-fluorine complexes. The latter effectively act as traps for the excess self-interstitials that would normally cause boron transient enhanced diffusion. Instead, fluorine-boron interactions are marginal and do not play any significant role. Our results are also consistent with other observations such as native-defect trapping and bubble formation. (Read more)
- 94. Microelectron. Reliability 45, 71 (2005) , “A comprehensive model of PMOS NBTI degradation ”,Negative bias temperature instability has become an important reliability concern for ultra-scaled Silicon IC technology with significant implications for both analog and digital circuit design. In this paper, we construct a comprehensive model for NBTI phenomena within the framework of the standard reaction–diffusion model. We demonstrate how to solve the reaction–diffusion equations in a way that emphasizes the physical aspects of the degradation process and allows easy generalization of the existing work. We also augment this basic reaction–diffusion model by including the temperature and field-dependence of the NBTI phenomena so that reliability projections can be made under arbitrary circuit operating conditions. (Read more)
- 95. J. Appl. Phys. 97, 053704 (2005) , “The role of nitrogen-related defects in high-k dielectric oxides: Density-functional studies”, J. L. Gavartin, A. L. Shluger, A. S. Foster, G. I. BersukerUsing ab initio density-functional total energy and molecular-dynamics simulations, we study the effects of various forms of nitrogen postdeposition anneal (PDA) on the electric properties of hafnia in the context of its application as a gate dielectric in field-effect transistors. We... (Read more)
- 96. Appl. Phys. Lett. 86, 143507 (2005) , “First-principles studies of the intrinsic effect of nitrogen atoms on reduction in gate leakage current through Hf-based high-k dielectrics”, N. Umezawa, K. Shiraishi, T. Ohno, H. Watanabe, T. Chikyow, K. Torii, K. Yamabe, K. Yamada, H. Kitajima, T. ArikadoThe atomistic effects of N atoms on the leakage current through HfO2 high-k gate dielectrics have been studied from first-principles calculations within the framework of a generalized gradient approximation (GGA). It has been found that the intrinsic effects of N atoms drastically... (Read more)
- 97. Appl. Phys. Lett. 87, 062105 (2005) , “Negative-U property of oxygen vacancy in cubic HfO2”, Y. P. Feng, A. T. L. Lim, M. F. LiOxygen vacancy in cubic HfO2 was investigated using first-principles calculation based on density functional theory and generalized gradient approximation. Five different charge states (V++, V+, V0, V–, and... (Read more)
- 98. Physica B 376-377, 358-361 (2006) , “Pulsed EPR studies of Phosphorus shallow donors in diamond and SiC”, J. Isoya, M. Katagiri, T. Umeda, S. Koizumi, H. Kanda, N. T. Son, A. Henry, A. Gali, E. Janz?nPhosphorus shallow donors having the symmetry lower than Td are studied by pulsed EPR. In diamond:P and 3C–SiC:P, the symmetry is lowered to D2d and the density of the donor wave function on the phosphorus atom exhibits a predominant p-character. In 4H–SiC:P with the site symmetry of... (Read more)
- 99. Phys. Rev. Lett. 96, 145501 (2006) , “Identification of the Carbon Antisite-Vacancy Pair in 4H-SiC”, T. Umeda, N. T. Son, J. Isoya, E. Janzn, T. Ohshima, N. Morishita, H. Itoh, A. Gali, M. BockstedteThe metastability of vacancies was theoretically predicted for several compound semiconductors alongside their transformation into the antisite-vacancy pair counterpart; however, no experiment to date has unambiguously confirmed the existence of antisite-vacancy pairs. Using electron paramagnetic resonance and first principles calculations we identify the SI5 center as the carbon antisite-vacancy pair in the negative charge state (CSiVC-) in 4H-SiC. We suggest that this defect is a strong carrier-compensating center in n-type or high-purity semi-insulating SiC. (Read more)SiC| ENDOR EPR Theory electron-irradiation optical-spectroscopy thermal-meas./anneal-exp.| -1 -2 1.0eV~ 13C 29Si C1h C3v Carbon Csi EI5/6 HEI1 HEI5/6 Nitrogen P6/7 SI5 Silicon Vc antisite bistable/metastable dangling-bond n-type pair(=2) semi-insulating vacancy .inp files: SiC/SI5_C1h SiC/SI5_80K SiC/SI5_100K | last update: Takashi Fukushima
- 100. Phys. Rev. Lett. 96, 55501 (2006) , “Divacancy in 4H-SiC”, N. T. Son, P. Carlsson, J. ul Hassan, E. Janz?n, T. Umeda, J. Isoya, A. Gali, M. Bockstedte, N. Morishita, T. Ohshima, H. ItohElectron paramagnetic resonance and ab initio supercell calculations suggest that the P6/P7 centers, which were previously assigned to the photoexcited triplet states of the carbon vacancy-antisite pairs in the double positive charge state, are related to the triplet ground... (Read more)
- 101. Phys. Rev. Lett. 97, 016102 (2006) , “Scaling and Universality of Roughening in Thermal Oxidation of Si(001)”, Hiroo Omi, Hiroyuki Kageshima, and Masashi UematsuBy analyzing atomic force microscopy images, we derive a continuum equation that quantitatively explains the roughening at the Si(001)-SiO2 interface during thermal oxidation at the temperature at 1200 °C in an Ar atmosphere containing a small fraction of O2. We also show... (Read more)
- 102. Phys. Rev. Lett. 97, 066101 (2006) , “Structure and Interconversion of Oxygen-Vacancy-Related Defects on Amorphous Silica”, Chin-Lung Kuo and Gyeong S. HwangAtomic structure and structural stability of neutral oxygen vacancies on amorphous silica are investigated using combined Monte Carlo and density functional calculations. We find that, unlike their bulk counterparts, the Si-Si dimer configuration of surface oxygen vacancies is likely to be unstable... (Read more)
- 103. Phys. Rev. Lett. 97, 116101 (2006) , “Oxygen Migration, Agglomeration, and Trapping: Key Factors for the Morphology of the Si-SiO2 Interface”, L. Tsetseris and S. T. PantelidesThe measured activation energies for oxide growth rates at the initial and late stages of oxidation of Si are 2 and 1.2 eV, respectively. These values imply that oxidation can proceed at temperatures much smaller than the 800 °C normally used to obtain devices with exceptionally smooth... (Read more)
- 104. Phys. Rev. Lett. 97, 135901 (2006) , “Vacancy-Assisted Diffusion in Silicon: A Three-Temperature-Regime Model”, Damien Caliste and Pascal PochetIn this Letter we report kinetic lattice Monte Carlo simulations of vacancy-assisted diffusion in silicon. We show that the observed temperature dependence for vacancy migration energy is explained by the existence of three diffusion regimes for divacancies. This characteristic has been rationalized... (Read more)
- 105. Phys. Rev. Lett. 97, 155901 (2006) , “Proton Diffusion Mechanism in Amorphous SiO2”, Julien Godet and Alfredo PasquarelloWe study proton diffusion in amorphous SiO2 from the atomic scale to the long-range percolative regime. Ab initio molecular dynamics suggest that the dominant atomic process consists in cross-ring interoxygen hopping assisted by network vibrations. A statistical analysis accounting... (Read more)
- 106. Phys. Rev. Lett. 97, 226401 (2006) , “Quasiparticle Corrections to the Electronic Properties of Anion Vacancies at GaAs(110) and InP(110)”, Magnus Hedström, Arno Schindlmayr, Günther Schwarz, and Matthias SchefflerWe propose a new method for calculating optical defect levels and thermodynamic charge-transition levels of point defects in semiconductors, which includes quasiparticle corrections to the Kohn-Sham eigenvalues of density-functional theory. Its applicability is demonstrated for anion vacancies at... (Read more)
- 107. Phys. Rev. Lett. 97, 255902 (2006) , “Atomistic Mechanism of Boron Diffusion in Silicon”, Davide De Salvador, Enrico Napolitani, Salvatore Mirabella, Gabriele Bisognin, Giuliana Impellizzeri, Alberto Carnera, and Francesco PrioloB diffuses in crystalline Si by reacting with a Si self-interstitial (I) with a frequency g and so forming a fast migrating BI complex that can migrate for an average length λ. We experimentally demonstrate that both g and λ strongly depend on the free hole... (Read more)
- 108. Phys. Rev. Lett. 97, 256602 (2006) , “Bistability-Mediated Carrier Recombination at Light-Induced Boron-Oxygen Complexes in Silicon”, Mao-Hua Du, Howard M. Branz, Richard S. Crandall, and S. B. ZhangA first-principles study of the BO2 complex in B-doped Czochralski Si reveals a defect-bistability-mediated carrier recombination mechanism, which contrasts with the standard fixed-level Shockley-Read-Hall model of recombination. An O2 dimer distant from B causes only weak... (Read more)
- 109. Phys. Rev. B 73, 033204 (2006) , “Electrical characterization of metastable carbon clusters in SiC: A theoretical study”, A. Gali, N. T. Son, E. JanznFirst-principles calculations carried out in 3C- and 4H-SiC show that small metastable carbon clusters can be created in irradiated SiC. The metastable carbon clusters possess occupation levels in the p-type as well as in the n-type 4H-SiC. Depending on the... (Read more)
- 110. Phys. Rev. B 73, 073302 (2006) , “Origin of Pb1 center at SiO2/Si(100) interface: First-principles calculations”, K. Kato, T. Yamasaki, T. UdaBased on first-principles calculations, we studied the generation behavior of Pb centers at SiO2/Si interfaces, especially for Pb1 centers, under oxidation of Si(100) surfaces. Pb1 centers were found to be formed... (Read more)
- 111. Phys. Rev. B 73, 075201 (2006) , “Electron paramagnetic resonance and theoretical studies of shallow phosphorous centers in 3C-, 4H-, and 6H-SiC”, N. T. Son, A. Henry, J. Isoya, M. Katagiri, T. Umeda, A. Gali, E. Janz?nContinuous-wave (cw) electron paramagnetic resonance (EPR) at both X-band and W-band frequencies, pulsed-EPR, and pulsed electron nuclear double resonance (ENDOR) were used to study phosphorus shallow donors in 3C-, 4H-, and 6H-SiC doped with phosphorus (P) during... (Read more)
- 112. Phys. Rev. B 73, 115207 (2006) , “First-principles study of migration mechanisms and diffusion of oxygen in zinc oxide”, Paul Erhart and Karsten AlbeWe have performed density-functional theory calculations in conjunction with the climbing image nudged elastic band method in order to study the self-diffusion of oxygen in zinc oxide. To this end, we have derived the complete set of migration paths for vacancies as well as interstitials in wurtzite... (Read more)
- 113. Phys. Rev. B 73, 125205 (2006) , “First-principles study of native defects in anatase TiO2”, Sutassana Na-Phattalung, M. F. Smith, Kwiseon Kim, Mao-Hua Du, Su-Huai Wei, S. B. Zhang, and Sukit LimpijumnongNative point defects in anatase TiO2 are investigated by using first-principles pseudopotential calculations based on density-functional theory (DFT). Antisite defects, namely, Ti-antisite (TiO) and O-antisite (OTi), have high formation energies and are hence... (Read more)
- 114. Phys. Rev. B 73, 125206 (2006) , “Atomistic simulations on the thermal stability of the antisite pair in 3C- and 4H-SiC”, M. Posselt, F. Gao, W. J. WeberThe thermal stability of the first-neighbor antisite pair configurations in 3C- and 4H-SiC is investigated by atomic-level computer simulations. First, the structure and energetics of these defects are determined in order to check the accuracy of the interatomic potential employed. The results are... (Read more)
- 115. Phys. Rev. B 73, 161201(R) (2006) , “Thermally stable carbon-related centers in 6H-SiC: Photoluminescence spectra and microscopic models”, A. Mattausch, M. Bockstedte, O. Pankratov, J. W. Steeds, S. Furkert, J. M. Hayes, W. Sullivan, N. G. WrightRecent ab initio calculations [Mattausch et al., Phys. Rev. B 70, 235211 (2004)] of carbon clusters in SiC reveal a possible connection between the tricarbon antisite (C3)Si and the U photoluminescence center in 6H-SiC [Evans et al., Phys. Rev. B 66, 35204... (Read more)
- 116. Phys. Rev. B 73, 184105 (2006) , “Iron-oxygen vacancy defect association in polycrystalline iron-modified PbZrO3 antiferroelectrics: Multifrequency electron paramagnetic resonance and Newman superposition model analysis”, Hrvoje Metri, Rüdiger-A. Eichel, Klaus-Peter Dinse, Andrew Ozarowski, Johan van Tol, Louis Claude Brunel, Hans Kungl, Michael J. Hoffmann, Kristin A. Schönau, Michael Knapp, and Hartmut FuessBy utilizing multifrequency electron paramagnetic resonance (EPR) spectroscopy, the iron functional center in Fe3+-modified polycrystalline lead zirconate (PbZrO3) was studied. The single phase polycrystalline sample remained orthorhombic and antiferroelectric down to 20 K as... (Read more)
- 117. Phys. Rev. B 73, 193202 (2006) , “First-principles study of point defects in rutile TiO2–x”, Eunae Cho, Seungwu Han, Hyo-Shin Ahn, Kwang-Ryeol Lee, Seong Keun Kim, and Cheol Seong HwangWe report our first-principles results on point defects in TiO2 in the rutile phase. Both the oxygen vacancy and titanium interstitial are considered. The size effect of the supercell has been examined and the localized state associated with the oxygen vacancy turns out to be sensitive to... (Read more)
- 118. Phys. Rev. B 73, 193203 (2006) , “Theoretical study of the phosphorus vacancy in ZnGeP2”, Xiaoshu Jiang, M. S. Miao, and Walter R. L. LambrechtFirst-principles calculations are presented for the phosphorus vacancy VP in ZnGeP2, using full-potential linearized muffin-tin orbital supercell local density functional theory calculations. We find the VP to have a high energy of formation compared to... (Read more)
- 119. Phys. Rev. B 73, 195206 (2006) , “Relaxations and bonding mechanism in Hg1–xCdxTe with mercury vacancy defect: First-principles study”, L. Z. Sun, Xiaoshuang Chen, Y. L. Sun, X. H. Zhou, Zh. J. Quan, He Duan, and Wei LuThe structural and electronic properties of the mercury vacancy defect in Hg1–xCdxTe have been studied by combining the full-potential linear augmented plane wave and plane-wave pseudopotential method base on the density functional theory. Structural... (Read more)
- 120. Phys. Rev. B 73, 205203 (2006) , “First-principles study of intrinsic point defects in ZnO: Role of band structure, volume relaxation, and finite-size effects”, Paul Erhart, Karsten Albe, and Andreas KleinDensity-functional theory (DFT) calculations of intrinsic point defect properties in zinc oxide were performed in order to remedy the influence of finite-size effects and the improper description of the band structure. The generalized gradient approximation (GGA) with empirical self-interaction... (Read more)
- 121. Phys. Rev. B 73, 235206 (2006) , “LSDA+U study of cupric oxide: Electronic structure and native point defects”, Dangxin Wu and Qiming ZhangA first-principles study on strongly correlated monoclinic cupric oxide CuO has been performed by using the LSDA+U method. The optimized structural parameters of the crystal CuO are in good agreement with the experimental data. The electronic structures and magnetic properties calculated from the... (Read more)
- 122. Phys. Rev. B 73, 235209 (2006) , “Temperature dependence of the vibrational spectrum of a Li-OH complex in ZnO: Infrared absorption experiments and theory”, Kevin R. Martin, Philip Blaney, Gang Shi, Michael Stavola, and W. Beall FowlerConsiderable interest has developed in the potential use of II–VI oxides as electronic and optical materials. In several cases, alkali atoms have been suggested as dopants. We report on the theoretical and experimental investigation of infrared and vibrational properties of a Li-OH complex in... (Read more)
- 123. Phys. Rev. B 73, 235211 (2006) , “Ab initio calculations for the interconversion of optically active defects in amorphous silica”, M. M. G. Alemany and James R. ChelikowskyUsing ab initio calculations on clusters, we have identified a new reaction path between the dicoordinated silicon atom defect and the paramagnetic Egamma[prime]" align="middle"> center in amorphous silica. Under ionizing irradiation, the dicoordinated silicon atom... (Read more)
- 124. Phys. Rev. B 73, 235213 (2006) , “Donor-vacancy complexes in Ge: Cluster and supercell calculations”, J. Coutinho, S. Öberg, V. J. B. Torres, M. Barroso, R. Jones, and P. R. BriddonWe present a comprehensive spin-density functional modeling study of the structural and electronic properties of donor-vacancy complexes (PV, AsV, SbV, and BiV) in Ge crystals. Special attention is paid to spurious results which are related to the choice of the boundary... (Read more)
- 125. Phys. Rev. B 73, 245203 (2006) , “Diffusion mechanisms for silicon di-interstitials”, Yaojun A. Du, Richard G. Hennig, and John W. WilkinsTight-binding molecular dynamics and density-functional simulations on silicon seeded with a di-interstitial reveal its detailed diffusion mechanisms. The lowest-energy di-interstitial performs a translation/rotation diffusion-step with a barrier of 0.3 eV and a prefactor of 11 THz followed by a... (Read more)
- 126. Phys. Rev. B 73, 245210 (2006) , “First-principles investigation of a bistable boron-oxygen interstitial pair in Si”, A. Carvalho, R. Jones, M. Sanati, S. K. Estreicher, J. Coutinho, and P. R. BriddonLocal density functional calculations are used to predict and compare the properties of the two distinct interstitial boron-interstitial oxygen (BiOi) complexes recently reported in the literature. The electronic and free energies, as well as the small... (Read more)
- 127. Phys. Rev. B 73, 245211 (2006) , “Hydrogen and muonium in diamond: A path-integral molecular dynamics simulation”, Carlos P. Herrero, Rafael Ramírez, and Eduardo R. HernándezIsolated hydrogen, deuterium, and muonium in diamond have been studied by path-integral molecular dynamics simulations in the canonical ensemble. Finite-temperature properties of these point defects were analyzed in the range from 100 to 800 K. Interatomic interactions were modeled by a... (Read more)
- 128. Phys. Rev. B 73, 245415 (2006) , “Ab initio study of nitrogen and boron substitutional impurities in single-wall SiC nanotubes”, A. GaliSilicon carbide nanotubes have a great potential for application in chemical sensors in harsh environment or in biological sensors. It is of interest to explore the electronic properties of these nanotubes, and how those are modified in the presence of impurities. It is well known that nitrogen and... (Read more)
- 129. Phys. Rev. B 74, 035202 (2006) , “Computation of the Stark effect in P impurity states in silicon”, A. Debernardi, A. Baldereschi, and M. FanciulliWe compute within the effective-mass theory and without adjustable parameters the Stark effect for shallow P donors in Si with anisotropic band structure. Valley-orbit coupling is taken into account in a nonperturbative way and scattering effects of the impurity core are included to properly... (Read more)
- 130. Phys. Rev. B 74, 035205 (2006) , “Mechanisms of arsenic clustering in silicon”, F. F. Komarov, O. I. Velichko, V. A. Dobrushkin, and A. M. MironovA model of arsenic clustering in silicon is proposed and analyzed. The main feature of the proposed model is the assumption that negatively charged arsenic complexes play a dominant role in the clustering process. To confirm this assumption, electron density and concentration of impurity atoms... (Read more)
- 131. Phys. Rev. B 74, 035210 (2006) , “Density-functional electronic structure calculations for native defects and Cu impurities in CdS”, Kazume Nishidate, Takuya Sato, Yuta Matsukura, Mamoru Baba, and Masayuki HasegawaWe performed density-functional electronic structure calculations for wurtzite CdS with native defects and Cu impurity. We investigate formation energies and ionization levels of the defects in various charge states. Our results reveal that the S vacancy is a double donor with the strongly localized... (Read more)
- 132. Phys. Rev. B 74, 045202 (2006) , “Effects of cation d states on the structural and electronic properties of III-nitride and II-oxide wide-band-gap semiconductors”, Anderson Janotti, David Segev, and Chris G. Van de WalleUsing first-principles methods based on density functional theory within the local density approximation (LDA) we calculate the structural and electronic properties of wurtzite MgO, ZnO, and CdO, and discuss their similarities and dissimilarities with the corresponding Group-III nitrides AlN, GaN,... (Read more)
- 133. Phys. Rev. B 74, 045213 (2006) , “Hyperfine interaction and magnetoresistance in organic semiconductors”, Y. Sheng, T. D. Nguyen, G. Veeraraghavan, Ö. Mermer, M. Wohlgenannt, S. Qiu, and U. ScherfWe explore the possibility that hyperfine interaction causes the recently discovered organic magnetoresistance (OMAR) effect. We deduce a simple fitting formula from the hyperfine Hamiltonian that relates the saturation field of the OMAR traces to the hyperfine coupling constant. We compare the... (Read more)
- 134. Phys. Rev. B 74, 045217 (2006) , “Vacancy clustering and diffusion in silicon: Kinetic lattice Monte Carlo simulations”, Benjamin P. Haley, Keith M. Beardmore, and Niels Grønbech-JensenDiffusion and clustering of lattice vacancies in silicon as a function of temperature, concentration, and interaction range are investigated by kinetic lattice Monte Carlo simulations. It is found that higher temperatures lead to larger clusters with shorter lifetimes on average, which grow by... (Read more)
- 135. Phys. Rev. B 74, 064105 (2006) , “Defect formation in LaGa(Mg,Ni)O3–δ: A statistical thermodynamic analysis validated by mixed conductivity and magnetic susceptibility measurements”, E. N. Naumovich, V. V. Kharton, A. A. Yaremchenko, M. V. Patrakeev, D. G. Kellerman, D. I. Logvinovich, and V. L. KozhevnikovA statistical thermodynamic approach to analyze defect thermodynamics in strongly nonideal solid solutions was proposed and validated by a case study focused on the oxygen intercalation processes in mixed-conducting LaGa0.65Mg0.15Ni0.20O3–δ... (Read more)
- 136. Phys. Rev. B 74, 064116 (2006) , “Charge centers in CaF2: Ab initio calculation of elementary physical properties”, M. Letz and L. ParthierCharge centers in ionic crystals provide a channel for elementary interaction between electromagnetic radiation and the lattice. We calculate the electronic ground-state energies which are needed to create a charge center—namely an F center and an H center. In good agreement with... (Read more)
- 137. Phys. Rev. B 74, 081201 (2006) , “Design of shallow acceptors in ZnO: First-principles band-structure calculations”, Jingbo Li, Su-Huai Wei, Shu-Shen Li, and Jian-Bai Xiap-type doping is a great challenge for the full utilization of ZnO as short-wavelength optoelectronic material. Due to a large electronegative characteristic of oxygen, the ionization energy of acceptors in ZnO is usually too high. By analyzing the defect wave-function character, we propose... (Read more)
- 138. Phys. Rev. B 74, 104303 (2006) , “Nitrogen-vacancy center in diamond: Model of the electronic structure and associated dynamics”, N. B. Manson, J. P. Harrison, and M. J. SellarsSymmetry considerations are used in presenting a model of the electronic structure and the associated dynamics of the nitrogen-vacancy center in diamond. The model accounts for the occurrence of optically induced spin polarization, for the change of emission level with spin polarization and for new... (Read more)
- 139. Phys. Rev. B 74, 113301 (2006) , “Reactions of excess hydrogen at a Si(111) surface with H termination: First-principles calculations”, L. Tsetseris, S. T. PantelidesHydrogen reactions with silicon substrates is an established technique for the study and control of surface morphology. Here, we report the results of first-principles calculations on the trapping and depassivation reactions involving excess hydrogen (x-H) at a fully H-passivated Si(111) surface. We find that x-H atoms can depassivate Si-H bonds with a small barrier of 0.8 eV, or they can get trapped in very stable configurations that comprise of a dihydride and a vicinal Si-H bond. Desorption of H2 molecules from these complexes has an activation energy of 1.68 eV, which can account for pertinent experimental data. We discuss also the effect of strain on the possibility of altering the x-H surface profile. (Read more)
- 140. Phys. Rev. B 74, 115201 (2006) , “Magnetic circular dichroism spectra in a II-VI diluted magnetic semiconductor Zn1–xCrxTe: First-principles calculations”, Hongming Weng, Jinming Dong, Tomoteru Fukumura, Masashi Kawasaki, and Yoshiyuki KawazoeThe absorption and magnetic circular dichroism (MCD) spectra of Zn1–xCrxTe for x=0.0625, 0.25, and 1.0 are studied using the first-principles method. The calculated MCD spectra are found to be well consistent with the experimental measurement at the... (Read more)
- 141. Phys. Rev. B 74, 121201 (2006) , “Prediction of the anomalous fluorine-silicon interstitial pair diffusion in crystalline silicon”, Scott A. Harrison, Thomas F. Edgar, and Gyeong S. HwangWe propose a diffusion pathway for a fluorine-silicon interstitial pair (F-Sii) in silicon based on extensive first-principles density functional calculations. We find the F-Sii pair to be most stable in the singly positive charge state under intrinsic conditions and can exist... (Read more)
- 142. Phys. Rev. B 74, 125203 (2006) , “Density functional theory of structural transformations of oxygen-deficient centers in amorphous silica during hole trapping: Structure and formation mechanism of the Egamma[prime]" align="middle"> center”, T. Uchino and T. YokoWe investigate the hole trapping process of a neutral oxygen vacancy in amorphous silicon dioxide (a-SiO2) using cluster calculations based on the density functional theory (DFT) method. We show that trapping a hole at a neutral oxygen vacancy leads to the formation of several... (Read more)
- 143. Phys. Rev. B 74, 144106 (2006) , “Density functional theory calculation of the DI optical center in SiC”, T. A. G. Eberlein, R. Jones, S. Öberg, and P. R. BriddonThe DI center is a prominent defect which is detected in as-grown or irradiated SiC. It is unusual in that its intensity grows with heat treatments and survives anneals of 1700 °C. It has been assigned recently to either a close-by antisite pair or to the close-by antisite... (Read more)
- 144. Phys. Rev. B 74, 153403 (2006) , “Doping and the unique role of vacancies in promoting the magnetic ground state in carbon nanotubes and C60 polymers”, Antonis N. Andriotis, R. Michael Sheetz, and Madhu MenonThe role of various types of defects in establishing the magnetic properties of the C60-based polymers and the single-wall carbon nanotubes is investigated. Comparing the role of carbon vacancies, and that of substitutional impurity atoms X (X=N, B, O, Si, P, and S) in... (Read more)
- 145. Phys. Rev. B 74, 155201 (2006) , “Crystal-field theory of Co2+ in doped ZnO”, R. O. Kuzian, A. M. Daré, P. Sati, and R. HaynWe present a crystal-field theory of transition-metal impurities in semiconductors in a trigonally distorted tetrahedral coordination. We develop a perturbative scheme to treat covalency effects within the weak ligand field case (Coulomb interaction dominates over one-particle splitting) and apply... (Read more)
- 146. Phys. Rev. B 74, 155205 (2006) , “Ab initio studies of the electronic structure of defects in PbTe”, Salameh Ahmad, S. D. Mahanti, Khang Hoang, and M. G. KanatzidisUnderstanding the detailed electronic structure of deep defect states in narrow band-gap semiconductors has been a challenging problem. Recently, self-consistent ab initio calculations within density functional theory using supercell models have been successful in tackling this problem. In... (Read more)
- 147. Phys. Rev. B 74, 155208 (2006) , “Vibrational spectra of vitreous germania from first-principles”, Luigi Giacomazzi, P. Umari, and Alfredo PasquarelloWe report on a first-principles investigation of the structural and vibrational properties of vitreous germania (v-GeO2). Our work focuses on a periodic model structure of 168 atoms, but three smaller models are also studied for comparison. We first carry out a detailed structural... (Read more)
- 148. Phys. Rev. B 74, 155425 (2006) , “Ab initio study of native defects in SiC nanotubes”, R. J. Baierle, P. Piquini, L. P. Neves, and R. H. MiwaSpin-polarized density functional theory is used to investigate the electronic and structural properties of vacancies and antisites in zigzag, armchair, and chiral SiC nanotubes. Antisites present lower formation energies compared to vacancies, introducing an empty electronic level close to the... (Read more)
- 149. Phys. Rev. B 74, 165116 (2006) , “Density-functional-theory calculations for the silicon vacancy”, A. F. WrightThe atomic configurations and formation energies of a silicon vacancy in the +2, +1, 0, −1, and −2 charge states have been computed using density-functional theory with norm-conserving pseudopotentials and a plane wave basis. Calculations were performed in simple cubic supercells using... (Read more)
- 150. Phys. Rev. B 74, 165404 (2006) , “Density functional study of gold atoms and clusters on a graphite (0001) surface with defects”, Jaakko Akola and Hannu HäkkinenAdsorption of gold atoms and clusters (N=6) on a graphite (0001) surface with defects has been studied using density functional theory. In addition to perfect graphite (0001), three types of surface defects have been considered: a surface vacancy (hole), a pyridinelike defect comprising three... (Read more)
- 151. Phys. Rev. B 74, 174101 (2006) , “First-principles study of the intrinsic defects in PbFCl”, Bo Liu, Zeming Qi, and Chaoshu ShiFirst-principles pseudopotential calculations have been performed to investigate intrinsic defects including vacancies, interstitials, antisite defects, as well as Schottky and Frenkel defects in PbFCl crystals. For the isolated vacancies and interstitials, their formation energies are critically... (Read more)
- 152. Phys. Rev. B 74, 174115 (2006) , “Modeling of damage generation mechanisms in silicon at energies below the displacement threshold”, Iván Santos, Luis A. Marqués, and Lourdes PelazWe have used molecular dynamics simulation techniques to study the generation of damage in Si within the low-energy deposition regime. We have demonstrated that energy transfers below the displacement threshold can produce a significant amount of damage, usually neglected in traditional radiation... (Read more)
- 153. Phys. Rev. B 74, 174407 (2006) , “Superexchange in dilute magnetic dielectrics: Application to (Ti,Co)O2”, K. Kikoin and V. FleurovWe extend the model of ferromagnetic superexchange in dilute magnetic semiconductors to the ferromagnetically ordered highly insulating compounds (dilute magnetic dielectrics). The intrinsic ferromagnetism without free carriers is observed in oxygen-deficient films of anatase TiO2 doped... (Read more)
- 154. Phys. Rev. B 74, 184108 (2006) , “Interpreting Mössbauer spectra reflecting an infinite number of sites: An application to Fe1−xSi synthesized by pulsed laser annealing”, A. Falepin, S. Cottenier, C. M. Comrie, and A. VantommeWe present a study on the interpretation of conversion electron Mössbauer spectra reflecting an infinite number of sites, in casu Mössbauer spectroscopy on Fe1−xSi layers on Si, synthesized by pulsed laser annealing. These spectra display a broad... (Read more)
- 155. Phys. Rev. B 74, 184117 (2006) , “Ab initio thermodynamic properties of point defects and O-vacancy diffusion in Mg spinels”, Zbigniew odziana and Jacek PiechotaWe report ab initio plane wave density functional theory studies of thermodynamic properties of isolated cation substitutions and oxygen vacancies in magnesium spinel, MgAl2O4. The formation enthalpy of Ca, Cu, and Zn substitutions of Mg cation indicate that transition... (Read more)
- 156. Phys. Rev. B 74, 193405 (2006) , “Role of charge in destabilizing AlH4 and BH4 complex anions for hydrogen storage applications: Ab initio density functional calculations”, A. J. Du, Sean C. Smith, and G. Q. LuNaAlH4 and LiBH4 are potential candidate materials for mobile hydrogen storage applications, yet they have the drawback of being highly stable and desorbing hydrogen only at elevated temperatures. In this letter, ab initio density functional theory calculations reveal... (Read more)
- 157. Phys. Rev. B 74, 195128 (2006) , “First-principles calculations of native defects in tin monoxide”, A. Togo, F. Oba, I. Tanaka, and K. TatsumiThe formation energies and electronic structure of native defects in tin monoxide are investigated by first-principles calculations. Equilibrium defect concentrations, which are obtained using the calculated formation energies and charge neutrality, indicate that the tin vacancy is the dominant... (Read more)
- 158. Phys. Rev. B 74, 195202 (2006) , “Interstitial-mediated mechanisms of As and P diffusion in Si: Gradient-corrected density-functional calculations”, Scott A. Harrison, Thomas F. Edgar, and Gyeong S. HwangGradient-corrected density-functional calculations are used to determine the structure, stability, and diffusion of arsenic-interstitial and phosphorus-interstitial pairs in the positive, neutral, and negative charge states. For both cases, our calculations show that the neutral pair will be... (Read more)
- 159. Phys. Rev. B 74, 205202 (2006) , “Structural model of amorphous silicon annealed with tight binding”, N. Bernstein, J. L. Feldman,, and M. FornariWe present a model of amorphous silicon generated by extensive annealing of a continuous random network structure using a molecular dynamics simulation with forces computed by a tight-binding total energy method. We also produce a refined model by relaxing the annealed model using density functional... (Read more)
- 160. Phys. Rev. B 74, 205207 (2006) , “Charge-dependent migration pathways for the Ga vacancy in GaAs”, Fedwa El-Mellouhi, Norm, and MousseauUsing a combination of the local-basis ab initio program SIESTA and the activation-relaxation technique we study the diffusion mechanisms of the gallium vacancy in GaAs. Vacancies are found to diffuse to the second neighbor using two different mechanisms, as well as to the first and fourth... (Read more)
- 161. Phys. Rev. B 74, 205208 (2006) , “Formation energies, binding energies, structure, and electronic transitions of Si divacancies studied by density functional calculations”, R. R. Wixom and A. F. WrightAtomic configurations, formation energies, electronic transition energies, and binding energies of the silicon divacancy in the +1, 0, −1, and −2 charge states were obtained from density functional theory calculations. The calculations were performed using the local density approximation... (Read more)
- 162. Phys. Rev. B 74, 205324 (2006) , “Surface smoothness of plasma-deposited amorphous silicon thin films: Surface diffusion of radical precursors and mechanism of Si incorporation”, Mayur S. Valipa, Tamas Bakos, and Dimitrios MaroudasWe present a detailed analysis of the fundamental atomic-scale processes that determine the surface smoothness of hydrogenated amorphous silicon (a-Si:H) thin films. The analysis is based on a synergistic combination of molecular-dynamics (MD) simulations of radical precursor migration on surfaces... (Read more)
- 163. Phys. Rev. B 74, 214105 (2006) , “Atomistic simulations of radiation-induced defect formation in spinels: MgAl2O4, MgGa2O4, and MgIn2O4”, D. Bacorisen, Roger Smith, B. P. Uberuaga, K. E. Sickafus, J. A. Ball, and R. W. GrimesMolecular dynamics simulations of collision cascades were performed in three spinel oxides with varying inversion, namely normal magnesium aluminate, MgAl2O4, half-inverse magnesium gallate, MgGa2O4, and inverse magnesium indate,... (Read more)
- 164. Phys. Rev. B 74, 214109 (2006) , “Inversion of defect interactions due to ordering in Sr1−3x/2LaxTiO3 perovskites: An atomistic simulation study”, B. S. Thomas, N. A. Marks, and Peter HarrowellSr1−3x/2LaxTiO3 perovskites exhibit large variations in radiation resistance and A-site vacancy ordering, depending on x and thermal history. In this study we use a combination of ab initio and classical simulation techniques... (Read more)
- 165. Phys. Rev. B 74, 233201 (2006) , “Electronic and magnetic properties of V-doped anatase TiO2 from first principles”, Xiaosong Du, Qunxiang Li, Haibin Su, and Jinlong YangWe report a first-principles study on the geometric, electronic, and magnetic properties of V-doped anatase TiO2. The DFT+U (Hubbard coefficient) approach predicts semiconductor band structures for Ti1−xVxO2 (x=6.25% and... (Read more)
- 166. Phys. Rev. B 74, 235207 (2006) , “Vacancy-enhanced ferromagnetism in Fe-doped rutile TiO2”, Jun Chen, Paul Rulis, Lizhi Ouyang, S. Satpathy, and W. Y. ChingBased on a series of supercell density functional calculations of Fe-doped TiO2 both with and without O vacancy (VO), we show that VO plays an important role in determining the magnetic properties of the dilute magnetic semiconductors (DMS). Without... (Read more)
- 167. Phys. Rev. B 74, 235208 (2006) , “Theoretical study of the magnetism of Mn-doped ZnO with and without defects”, D. Iuan, B. Sanyal, and O. ErikssonWe calculate the exchange interaction parameters of a classical Heisenberg Hamiltonian for Mn-doped ZnO (Mn concentration between 5% and 20%) by an ab initio Korringa-Kohn-Rostoker coherent-potential-approximation method in the framework of density functional theory. A weak antiferromagnetic... (Read more)
- 168. Phys. Rev. B 74, 235209 (2006) , “Comparison of two methods for circumventing the Coulomb divergence in supercell calculations for charged point defects”, A. F. Wright and N. A. ModineDensity-functional-theory calculations were performed for the unrelaxed +2 Si vacancy and +2 self-interstitial utilizing periodic boundary conditions and two different methods—the uniform background charge method and the local moment counter charge method—for circumventing the divergence... (Read more)
- 169. Phys. Rev. B 74, 235301 (2006) , “Precursor mechanism for interaction of bulk interstitial atoms with Si(100)”, Xiao Zhang, Min Yu, Charlotte T. M. Kwok, Ramakrishnan Vaidyanathan, Richard D. Braatz, and Edmund G. SeebauerIn the same way that gases react with surfaces from above, solid-state point defects such as interstitial atoms can react from below. Little attention has been paid to this form of surface chemistry. Recent bulk self-diffusion measurements near the Si(100) surface have quantified Si interstitial... (Read more)
- 170. Phys. Rev. B 74, 235426 (2006) , “First-principles study of electronic properties of hydrogenated graphite”, A. Allouche and Y. FerroProgressive implantation of hydrogen atoms into graphite is modeled by first-principles density functional theory. The maximum H/C ratio was found at 53%, which is in good agreement with experimental results. The hydrogen trapping energy varies from −0.7 eV at very low concentrations to a... (Read more)
- 171. Phys. Rev. B 74, 241303(R) (2006) , “Enhancement of interactions between magnetic ions in semiconductors due to declustering”, A. Franceschetti, S. V. Barabash, J. Osorio-Guillen, A. Zunger, and M. van SchilfgaardeIt is often assumed that the exchange interaction between two magnetic ions in a semiconductor host depends only on the distance and orientation of the magnetic ions. Using first-principles electronic structure calculations of Mn impurities in GaAs, we show that the exchange interaction between two... (Read more)
- 172. Phys. Rev. B 74, 245212 (2006) , “Deactivation of nitrogen donors in silicon carbide”, F. Schmid, S. A. Reshanov, H. B. Weber, G. Pensl, M. Bockstedte, A. Mattausch, O. Pankratov, T. Ohshima, and H. ItohHexagonal SiC is either co-implanted with silicon (Si+), carbon (C+), or neon (Ne+) ions along with nitrogen (N+) ions or irradiated with electrons (e−) of 200 keV energy. During the subsequent annealing step at temperatures above 1450 ... (Read more)
- 173. Phys. Rev. B 74, 245216 (2006) , “Influence of excited states of a deep substitutional dopant on majority-carrier concentration in semiconductors”, Hideharu MatsuuraThe density (NA) and energy level (EA) of an acceptor in a p-type wide-band-gap semiconductor (e.g., SiC, GaN, and diamond) are determined by a least-squares fit of the charge neutrality equation to the temperature dependence of the hole... (Read more)
- 174. Phys. Rev. B 74, 245217 (2006) , “Donor levels for selected n-type dopants in diamond: A computational study of the effect of supercell size”, J. P. Goss, P. R. Briddon, and R. J. EyreComputational techniques are key predictive tools in the drive to engineer semiconductive materials. Diamond, intrinsically a wide band-gap insulator, can be made to semiconduct n-type by doping with phosphorus. However, the relatively deep level at Ec−0.6 eV... (Read more)
- 175. Phys. Rev. B 74, 245411 (2006) , “Vacancy defects and the formation of local haeckelite structures in graphene from tight-binding molecular dynamics”, Gun-Do Lee, C. Z. Wang, Euijoon Yoon, Nong-Moon Hwang, and K. M. HoThe dynamics of multivacancy defects in a graphene layer is investigated by tight-binding molecular dynamics simulations and by first principles calculation. The simulations show that four single vacancies in the graphene layer first coalesce into two double vacancies, each consisting of a... (Read more)
- 176. Phys. Rev. B 74, 245420 (2006) , “Energetics, structure, and long-range interaction of vacancy-type defects in carbon nanotubes: Atomistic simulations”, J. Kotakoski, A. V. Krasheninnikov,, and K. NordlundThe presence of vacancy clusters in carbon nanotubes has been assumed to explain the formation of carbon peapods and the difference between the experimentally measured and theoretical fracture strength of nanotubes. We use atomistic simulations at various levels of theory to study the... (Read more)
- 177. Microelectron. Reliability 46, 1 (2006) , “NBTI degradation: From physical mechanisms to modelling”,An overview of the evolution of transistor parameters under negative bias temperature instability stress conditions commonly observed in p-MOSFETs in recent technologies is presented. The physical mechanisms of the degradation as well as the different defects involved have been discussed according to a systematic set of experiments with different stress conditions. According to our findings, a physical model is proposed which could be used to more accurately predict the transistor degradation. Finally, based on our new present understanding, a new characterization methodology is proposed, which would open the way to a more accurate determination of parameter shifts and thus allowing implementing the degradation into design rules. (Read more)
- 178. J. Appl. Phys. 99, 011101 (2006) , “Degradation of hexagonal silicon-carbide-based bipolar devices”, M. Skowronski and S. HaOnly a few years ago, an account of degradation of silicon carbide high-voltage p-i-n diodes was presented at the European Conference on Silicon Carbide and Related Compounds (Kloster Banz, Germany, 2000). This report was followed by the intense effort of multiple groups... (Read more)
- 179. J. Appl. Phys. 99, 044105 (2006) , “Passivation of oxygen vacancy states in HfO2 by nitrogen”, K. Xiong, J. Robertson, and S. J. ClarkNitrogen is known to reduce leakage currents and charge trapping in high-dielectric-constant gate oxides such as HfO2. We show that this occurs because nitrogen, substituting for oxygen atoms next to oxygen vacancy sites, repels the occupied gap states due to the neutral and positively... (Read more)
- 180. J. Appl. Phys. 99, 054507 (2006) , “Characterization of HfSiON gate dielectrics using monoenergetic positron beams”, A. Uedono, K. Ikeuchi, T. Otsuka, K. Shiraishi, K. Yamabe, S. Miyazaki, N. Umezawa, A. Hamid, T. Chikyow, T. Ohdaira M. Muramatsu, R. Suzuki, S. Inumiya, S. Kamiyama, Y. Akasaka, Y. Nara, and K. YamadaThe impact of nitridation on open volumes in thin HfSiOx films fabricated on Si substrates by atomic layer deposition was studied using monoenergetic positron beams. For HfSiOx, positrons were found to annihilate from the trapped state due to open volumes which... (Read more)
- 181. J. Appl. Phys. 99, 113506 (2006) , “Theoretical properties of the N vacancy in p-type GaN(Mg,H) at elevated temperatures”, S. M. Myers, A. F. Wright, M. Sanati, and S. K. EstreicherThe elevated-temperature properties of the N vacancy in Mg-doped, p-type GaN containing H were modeled using atomic-configuration energies and phonon densities of states obtained with density-functional theory. This study encompassed both equilibrium thermodynamics and the rates of diffusion... (Read more)
- 182. J. Appl. Phys. 100, 023512 (2006) , “Stacking faults and twin boundaries in fcc crystals determined by x-ray diffraction profile analysis”, Levente Balogh, Gábor Ribárik, and Tamás UngárA systematic procedure is developed to evaluate the density of planar defects together with dislocations and crystallite or subgrain size by x-ray line profile analysis in fcc crystals. Powder diffraction patterns are numerically calculated by using the DIFFAX software for intrinsic and extrinsic... (Read more)
- 183. J. Appl. Phys. 100, 024510 (2006) , “Influence of iron contamination on the performances of single-crystalline silicon solar cells: Computed and experimental results”, S. Dubois, O. Palais, M. Pasquinelli, S. Martinuzzi, C. Jaussaud, and N. RondelIn this paper, the impact of iron contamination on the conversion efficiency of single-crystalline p-type silicon solar cells is investigated by means of the combination of numerical simulations and experimental data, taking into account the more recent results about the properties of iron in... (Read more)
- 184. J. Appl. Phys. 100, 034309 (2006) , “Critical size for defects in nanostructured materials”, Jagdish NarayanThis paper addresses some of the fundamental issues and critical advantages in reducing the grain size/feature size to the nanoscale regime. We find that as the grain size or feature size is reduced, there is a critical size below which the defect content can be reduced virtually to zero. This... (Read more)
- 185. J. Appl. Phys. 100, 063517 (2006) , “Extraction of Cu diffusivities in dielectric materials by numerical calculation and capacitance-voltage measurement”, Ki-Su Kim, Young-Chang Joo, Ki-Bum Kim, and Jang-Yeon KwonA rigorous method of obtaining the Cu diffusivities in various SiO2-based dielectric materials is proposed. The diffusion profile of Cu ions in a dielectric material is first simulated and the resulting flatband voltage shift (VFB) is compared with the experimental... (Read more)
- 186. J. Appl. Phys. 100, 113513 (2006) , “Theoretical study of nitrogen-doping effects on void formation processes in silicon crystal growth”, Hiroyuki Kageshima, Akihito Taguchi, and Kazumi WadaNitrogen-doping effects in silicon crystal growth have been theoretically studied using thermodynamical simulation based on first-principles calculation results. The results show that the densities of various complexes are determined in the balance between the enthalpy effects and the entropy... (Read more)
- 187. J. Appl. Phys. 100, 123108 (2006) , “Density functional theory investigation of N interstitial migration in GaN”, R. R. Wixom and A. F. WrightUsing density-functional total energy calculations, we investigated N interstitial migration in GaN. Two migration paths were considered. The first path confines motion to a single c-plane of the lattice, while the second path involves movement both perpendicular and parallel to the... (Read more)
- 188. Appl. Phys. Lett. 88, 012101 (2006) , “The structure of the SiO2/Si(100) interface from a restraint-free search using computer simulations”, Dominik Fischer, Alessandro Curioni, Salomon Billeter, and Wanda AndreoniThe structure of the interface between SiO2 and Si(100) is investigated using the replica-exchange method driven by classical molecular dynamics simulations based on ab initio-derived interatomic potentials. Abrupt interfaces are shown to be unstable, whereas a substoichiometric... (Read more)
- 189. Appl. Phys. Lett. 88, 082113 (2006) , “Effect of charge on the movement of dislocations in SiC”, T. A. G. Eberlein, R. Jones, A. T. Blumenau, S. ?berg, P. R. BriddonSiC bipolar devices show a degradation under forward-biased operation which has been linked with a current induced motion of one of the two glide dislocations having either Si or C core atoms. We have carried out calculations of the core structures and dynamics of partial dislocations in 3C and... (Read more)
- 190. Appl. Phys. Lett. 88, 091919 (2006) , “Calculation of deep carrier traps in a divacancy in germanium crystals”, J. Coutinho, V. J. B. Torres, R. Jones, A. Carvalho, S. berg, P. R. BriddonWe present an ab initio density functional study on the electronic structure and electrical properties of divacancies in Ge. Although suffering essentially different Jahn-Teller distortions when compared to the analogous defect in Si, the relative location of the electrical levels in the gap... (Read more)
- 191. Appl. Phys. Lett. 88, 153518 (2006) , “Negative bias temperature instability mechanism: The role of molecular hydrogen”, Anand T. Krishnan, Srinivasan Chakravarthi, Paul Nicollian, Vijay Reddy, and Srikanth KrishnanThe role of dimerization of atomic hydrogen to give molecular hydrogen in determining negative bias temperature instability (NBTI) kinetics is explored analytically. The time dependency of NBTI involving molecular hydrogen was found to obey a power law with a slope of 1/6, as opposed to the 1/4... (Read more)
- 192. Appl. Phys. Lett. 88, 162107 (2006) , “Physical origin of threshold voltage problems in polycrystalline silicon/HfO2 gate stacks”, Dae Yeon Kim, Joongoo Kang, and K. J. ChangBased on theoretical calculations, we find that at p+ polycrystalline silicon (poly-Si)/HfO2 gates, Si interstitials are easily migrated from the electrode, forming Hf–Si bonds with a charge transfer to the electrode, and the resulting interface dipole raises the Fermi level... (Read more)
- 193. Appl. Phys. Lett. 88, 171912 (2006) , “Impact of nitridation on open volumes in HfSiOx studied using monoenergetic positron beams”, A. Uedono, K. Ikeuchi, T. Otsuka, K. Yamabe, K. Eguchi, M. Takayanagi, T. Ohdaira, M. Muramatsu, R. Suzuki, A. S. Hamid, T. ChikyowThe effects of nitridation on open volumes in thin HfSiOx films fabricated by metal-organic chemical vapor deposition were studied using monoenergetic positron beams. It was found that positrons were annihilated from the trapped state by open volumes that exist intrinsically in... (Read more)
- 194. Appl. Phys. Lett. 88, 182903 (2006) , “Effects of Al addition on the native defects in hafnia”, Q. Li, K. M. Koo, W. M. Lau, P. F. Lee, J. Y. Dai, Z. F. Hou, X. G. GongTwo occupied native defect bands are experimentally detected in pure HfO2. The density of states of band one in the middle of the band gap reduces drastically with the Al addition, while that of band two slightly above the valence-band maximum remains rather unaffected. We attribute the... (Read more)
- 195. Appl. Phys. Lett. 88, 201918 (2006) , “Diffusion of zinc vacancies and interstitials in zinc oxide”, Paul Erhart and Karsten AlbeThe self-diffusion coefficient of zinc in ZnO is derived as a function of the chemical potential and Fermi level from first-principles calculations. Density functional calculations in combination with the climbing image-nudged elastic band method are used in order to determine migration barriers for... (Read more)
- 196. Appl. Phys. Lett. 88, 253504 (2006) , “Single silicon vacancy-oxygen complex defect and variable retention time phenomenon in dynamic random access memories”, T. Umeda, K. Okonogi, K. Ohyu, S. Tsukada, K. Hamada, S. Fujieda, and Y. MochizukiThe variable retention time phenomenon has recently been highlighted as an important issue in dynamic random access memory (DRAM) technology. Based on electrically detected magnetic resonance and simulation studies, we suggest that a single Si vacancy-oxygen complex defect is responsible for this... (Read more)
- 197. Appl. Phys. Lett. 89, 053511 (2006) , “Density functional theory study of deep traps in silicon nitride memories”, Max Petersen and Yakov RoizinUsing density functional theory, the interaction of hydrogen with a nitrogen vacancy in -Si3N4 is investigated. A single H atom was found to be energetically favorable over non- and doubly protonated vacancies. The traps composed of excess silicon and hydrogen have negative... (Read more)
- 198. Appl. Phys. Lett. 89, 082908 (2006) , “Negative oxygen vacancies in HfO2 as charge traps in high-k stacks”, J. L. Gavartin, D. Muñoz Ramo, A. L. Shluger, G. Bersuker, and B. H. LeeThe optical excitation and thermal ionization energies of oxygen vacancies in m-HfO2 are calculated using a non-local density functional theory with atomic basis sets and periodic supercell. The thermal ionization energies of negatively charged V– and... (Read more)
- 199. Appl. Phys. Lett. 89, 112107 (2006) , “Determination of the concentration of recombination centers in thin asymmetrical p-n junctions from capacitance transient spectroscopy”, Juan A. Jiménez Tejada, Pablo Lara Bullejos, Juan A. López Villanueva, Francisco M. Gómez-Campos, Salvador Rodríguez-Bolívar, and M. Jamal DeenRecombination centers in thin asymmetrical p-n junctions were analyzed in the context of capacitance transient experiments. The combined effect of the thin low-doped region of the junction and the nonzero value of the occupation factor of the recombination center in the depletion layer... (Read more)
- 200. Appl. Phys. Lett. 89, 142914 (2006) , “Defect passivation in HfO2 gate oxide by fluorine”, K. Tse and J. RobertsonThe authors have calculated that fluorine substituting for oxygen gives no gap states in HfO2. This accounts for the good passivation of oxygen vacancies by F seen experimentally. Bonding arguments are used to account for why F may be the most effective passivant in ionic oxides such as... (Read more)
- 201. Appl. Phys. Lett. 89, 151923 (2006) , “Variable core model and the Peierls stress for the mixed (screw-edge) dislocation”, Vlado A. Lubarda and Xanthippi MarkenscoffA variable core model of a moving crystal dislocation is proposed and used to derive an expression for the Peierls stress. The dislocation width varies periodically as a dislocation moves through the lattice, which leads to an expression for the Peierls stress in terms of the difference of the total... (Read more)
- 202. Appl. Phys. Lett. 89, 152904 (2006) , “First principles calculations of oxygen vacancy passivation by fluorine in hafnium oxide”, Wei Chen, Qing-Qing Sun, Shi-Jin Ding, David Wei Zhang, and Li-Kang WangThe fluorine incorporation into HfO2 with oxygen vacancies has been investigated using first principles calculations. The authors show that atomic fluorine can efficiently passivate the neutral oxygen vacancy with excess energies of 4.98 and 4.39 eV for threefold- and... (Read more)
- 203. Appl. Phys. Lett. 89, 202904 (2006) , “Effects of O vacancies and C doping on dielectric properties of ZrO2: A first-principles study”, Gargi Dutta, K. P. S. S. Hembram, G. Mohan Rao, and Umesh V. WaghmareThe authors determine electronic properties, structural stability, and dielectric response of zirconia (ZrO2) with oxygen vacancies (O vacancies) and carbon doping (C doping) using first-principles density functional theory calculations based on pseudopotentials and a plane wave basis.... (Read more)
- 204. Appl. Phys. Lett. 89, 262904 (2006) , “Oxygen vacancy in monoclinic HfO2: A consistent interpretation of trap assisted conduction, direct electron injection, and optical absorption experiments”, Peter Broqvist and Alfredo PasquarelloThe authors calculate energy levels associated with the oxygen vacancy in monoclinic HfO2 using a hybrid density functional which accurately reproduces the experimental band gap. The most stable charge states are obtained for varying Fermi level in the HfO2 band gap. To compare... (Read more)
- 205. Phys. Rev. Lett. 98, 015501 (2007) , “First-Principles Simulations of Boron Diffusion in Graphite”, I. Suarez-Martinez, A. A. El-Barbary, G. Savini, and M. I. HeggieBoron strongly modifies electronic and diffusion properties of graphite. We report the first ab initio study of boron interaction with the point defects in graphite, which includes structures, thermodynamics, and diffusion. A number of possible diffusion mechanisms of boron in graphite are... (Read more)
- 206. Phys. Rev. Lett. 98, 026101 (2007) , “Bonding at the SiC-SiO2 Interface and the Effects of Nitrogen and Hydrogen”, Sanwu Wang, S. Dhar, Shu-rui Wang, A. C. Ahyi, A. Franceschetti, J. R. Williams, L. C. Feldman,, and Sokrates T. PantelidesUnlike the Si-SiO2 interface, the SiC-SiO2 interface has large defect densities. Though nitridation has been shown to reduce the defect density, the effect of H remains an open issue. Here we combine experimental data and the results of first-principles calculations to... (Read more)
- 207. Phys. Rev. Lett. 98, 045501 (2007) , “Dopability, Intrinsic Conductivity, and Nonstoichiometry of Transparent Conducting Oxides”, Stephan Lany and Alex ZungerExisting defect models for In2O3 and ZnO are inconclusive about the origin of conductivity, nonstoichiometry, and coloration. We apply systematic corrections to first-principles calculated formation energies ΔH, and validate our theoretical defect model against... (Read more)
- 208. Phys. Rev. Lett. 98, 075503 (2007) , “Pseudoclimb and Dislocation Dynamics in Superplastic Nanotubes”, Feng Ding, Kun Jiao, Mingqi Wu, and Boris I. YakobsonPlastic relaxation of carbon nanotubes under tension and at high temperature is described in terms of dislocation theory and with atomistic computer simulations. It is shown how the glide of pentagon-heptagon defects and a particular type of their pseudoclimb, with the atoms directly breaking out of... (Read more)
- 209. Phys. Rev. Lett. 98, 115503 (2007) , “Oxygen Vacancy Clustering and Electron Localization in Oxygen-Deficient SrTiO3: LDA+U Study”, Do Duc Cuong, Bora Lee, Kyeong Mi Choi, Hyo-Shin Ahn, Seungwu Han, and Jaichan LeeWe find, using a local density approximation +Hubbard U method, that oxygen vacancies tend to cluster in a linear way in SrTiO3, a prototypical perovskite oxide, accompanied by strong electron localization at the 3d state of the nearby Ti transition metal ion. The vacancy... (Read more)
- 210. Phys. Rev. Lett. 98, 117202 (2007) , “Density-Functional Theory Study of Half-Metallic Heterostructures: Interstitial Mn in Si”, Hua Wu, Peter Kratzer, and Matthias SchefflerUsing density-functional theory within the generalized gradient approximation, we show that Si-based heterostructures with 1/4 layer δ doping of interstitial Mn (Mnint) are half-metallic. For Mnint concentrations of 1/2 or 1 layer, the states induced in the band... (Read more)
- 211. Phys. Rev. Lett. 98, 135506 (2007) , “Possible Approach to Overcome the Doping Asymmetry in Wideband Gap Semiconductors”, Yanfa Yan, Jingbo Li, Su-Huai Wei, and M. M. Al-JassimThe asymmetry doping problem has severely hindered the potential applications of many wideband gap (WBG) materials. Here, we propose a possible approach to overcome this long-standing doping asymmetry problem for WBG semiconductors. Our approach is based on the reduction of the ionization energies... (Read more)
- 212. Phys. Rev. Lett. 98, 137202 (2007) , “Magnetizing Oxides by Substituting Nitrogen for Oxygen”, I. S. Elfimov, A. Rusydi, S. I. Csiszar, Z. Hu, H. H. Hsieh, H.-J. Lin, C. T. Chen, R. Liang, and G. A. SawatzkyWe describe a possible pathway to new magnetic materials with no conventional magnetic elements present. The substitution of nitrogen for oxygen in simple nonmagnetic oxides leads to holes in N 2p states which form local magnetic moments. Because of the very large Hund's rule coupling of... (Read more)
- 213. Phys. Rev. Lett. 98, 196101 (2007) , “Oxygen Vacancies in High Dielectric Constant Oxide-Semiconductor Films”, Supratik Guha and Vijay NarayananWe provide evidence that the oxygen vacancy is a dominant intrinsic electronic defect in nanometer scaled hafnium oxide dielectric films on silicon, relevant to microelectronics technology. We demonstrate this by developing a general model for the kinetics of oxygen vacancy formation in... (Read more)
- 214. Phys. Rev. Lett. 98, 196802 (2007) , “Origin of Charge Density at LaAlO3 on SrTiO3 Heterointerfaces: Possibility of Intrinsic Doping”, Wolter Siemons, Gertjan Koster, Hideki Yamamoto, Walter A. Harrison, Gerald Lucovsky, Theodore H. Geballe, Dave H. A. Blank, and Malcolm R. BeasleyAs discovered by Ohtomo and Hwang, a large sheet charge density with high mobility exists at the interface between SrTiO3 and LaAlO3. Based on transport, spectroscopic, and oxygen-annealing experiments, we conclude that extrinsic defects in the form of oxygen vacancies... (Read more)
- 215. Phys. Rev. Lett. 98, 252501 (2007) , “Radioactive Decay Speedup at T=5 K: Electron-Capture Decay Rate of 7Be Encapsulated in C60”, T. Ohtsuki, K. Ohno, T. Morisato, T. Mitsugashira, K. Hirose, H. Yuki, and J. KasagiThe electron-capture (EC) decay rate of 7Be in C60 at the temperature of liquid helium (T=5 K) was measured and compared with the rate in Be metal at T=293 K. We found that the half-life of 7Be in endohedral C60... (Read more)
- 216. Phys. Rev. B 75, 014102 (2007) , “First-principles study of vacancy formation in hydroxyapatite”, Katsuyuki Matsunaga and Akihide KuwabaraFirst-principles plane-wave calculations were performed for hydroxyapatite (HAp) in order to investigate the electronic structure and vacancy formation mechanisms. The HAp unit cell contains PO4 tetrahedra and OH groups formed by covalent P-O and H-O bonds. Ca ions play a role for... (Read more)
- 217. Phys. Rev. B 75, 014111 (2007) , “Effects of vacancies on the properties of disordered ferroelectrics: A first-principles study”, L. Bellaiche, Jorge Íñiguez, Eric Cockayne, and B. P. BurtonA first-principles-based model is developed to investigate the influence of lead vacancies on the properties of the disordered Pb(Sc1/2Nb1/2)O3 (PSN) ferroelectric. Lead vacancies generate large, inhomogeneous, electric fields that reduce barriers between energy... (Read more)
- 218. Phys. Rev. B 75, 024109 (2007) , “Optical and EPR properties of point defects at a crystalline silica surface: Ab initio embedded-cluster calculations”, L. Giordano, P. V. Sushko, G. Pacchioni, and A. L. ShlugerWe have studied the structure and spectroscopic properties of the paramagnetic nonbridging oxygen hole center and of the Egamma[prime]" align="middle"> center at the hydroxylated silica surfaces using density functional theory and an embedded-cluster model. To investigate the... (Read more)
- 219. Phys. Rev. B 75, 024205 (2007) , “Mechanical strength and coordination defects in compressed silica glass: Molecular dynamics simulations”, Yunfeng Liang, Caetano R. Miranda, and Sandro ScandoloContrary to ordinary solids, which are normally known to harden by compression, the compressibility of SiO2 (silica) glass has a maximum at about 2–4 GPa and its mechanical strength shows a minimum around 10 GPa. At this pressure, the compression of silica glass undergoes a change... (Read more)
- 220. Phys. Rev. B 75, 033303 (2007) , “Excitons in silicon nanocrystallites: The nature of luminescence”, Eleonora Luppi, Federico Iori, Rita Magri, Olivia Pulci, Stefano Ossicini, Elena Degoli, and Valerio OlevanoThe absorption and emission spectra of silicon nanocrystals up to 1 nm diameter have been calculated within a first-principles framework. Our calculations include geometry optimization and the many-body effects induced by the creation of an electron-hole pair. Starting from hydrogenated silicon... (Read more)
- 221. Phys. Rev. B 75, 035210 (2007) , “Self- and foreign-atom diffusion in semiconductor isotope heterostructures. I. Continuum theoretical calculations”, H. BrachtDopant diffusion experiments in semiconductors yield the mobility of the element of interest and information about the possible mechanisms of atomic diffusion. In many cases the diffusion is described on the basis of Fick's law of diffusion, but this treatment is often too simple. In this paper,... (Read more)
- 222. Phys. Rev. B 75, 035211 (2007) , “Self- and foreign-atom diffusion in semiconductor isotope heterostructures. II. Experimental results for silicon”, H. Bracht, H. H. Silvestri, I. D. Sharp, and E. E. HallerWe report the diffusion of boron, arsenic, and phosphorus in silicon isotope multilayer structures at temperatures between 850 °C and 1100 °C. The diffusion of all dopants and self-atoms at a given temperature is modeled with the same setting of all native-point-defect-related parameters.... (Read more)
- 223. Phys. Rev. B 75, 035322 (2007) , “Ab initio study of electronic and magnetic properties of the C-codoped Ga1−xMnxN (100) surface”, Q. Wang, Q. Sun, and P. JenaFirst principles calculations based on gradient corrected density functional theory have been carried out to study the magnetic coupling between Mn atoms in pure and carbon doped Ga1−xMnxN thin films. We show that the ground state of Mn-doped GaN (100) thin... (Read more)
- 224. Phys. Rev. B 75, 045201 (2007) , “Ab initio models of amorphous Si1−xGex:H”, T. A. Abtew and D. A. DraboldWe study the structural, dynamical, and electronic properties of amorphous Si1−xGex:H alloys using first-principles local basis molecular dynamics techniques. The network topology and defects in the amorphous network have been analyzed. Structural changes,... (Read more)
- 225. Phys. Rev. B 75, 045211 (2007) , “Ab initio supercell calculations on aluminum-related defects in SiC”, A. Gali, T. Hornos, N. T. Son, E. Janzén, and W. J. ChoykeAb initio supercell calculations of the binding energies predict complex formation between aluminum and carbon interstitials in SiC. In high-energy implanted SiC aluminum acceptor can form very stable complexes with two carbon interstitials. We also show that carbon vacancy can be attached to... (Read more)
- 226. Phys. Rev. B 75, 073203 (2007) , “Diffusion mechanisms of native point defects in rutile TiO2: Ab initio total-energy calculations”, Hakim Iddir, Serdar Öüt, Peter Zapol, and Nigel D. BrowningThe structural energetics and diffusion mechanisms of the two most important point defects in rutile TiO2, the oxygen vacancy (VO) and the titanium interstitial (TiI), are examined using the ab initio pseudopotential total-energy method. The two... (Read more)
- 227. Phys. Rev. B 75, 073409 (2007) , “Why thermal H2 molecules adsorb on SiC(001)-c(4×2) and not on SiC(001)-(3×2) at room temperature”, Xiangyang Peng, Peter Krüger, and Johannes PollmannIn a recent experiment, Derycke et al. have made the exciting observation that H2 molecules readily adsorb dissociatively on the c(4×2) but not on the 3×2 surface of SiC(001) at room temperature. To unravel this spectacular reactivity difference, we have investigated a... (Read more)
- 228. Phys. Rev. B 75, 075202 (2007) , “Theoretical study of Li and Na as n-type dopants for diamond”, J. P. Goss and P. R. BriddonPhosphorus is the n-type dopant of choice for diamond, but results in a deep donor level and alternatives are being sought. One possibility is the incorporation of interstitial alkali metal impurities such as Li and Na. We present the results of density-functional calculations used to predict... (Read more)
- 229. Phys. Rev. B 75, 075206 (2007) , “Isotope dependence of the vibrational lifetimes of light impurities in Si from first principles”, D. West and S. K. EstreicherThe vibrational lifetimes of a range of H-related defects and interstitial O (Oi) in Si, including isotopic substitutions, are calculated from first principles as a function of temperature. The theoretical approach is explained in detail. The vibrational lifetimes of... (Read more)
- 230. Phys. Rev. B 75, 075312 (2007) , “Ferromagnetic to ferrimagnetic crossover in Cr-doped GaN nanohole arrays”, Q. Wang, Q. Sun, P. Jena, and Y. KawazoeUsing spin-polarized density-functional theory with exchange and correlation potential, approximated by both the generalized gradient approximation (GGA) and the GGA+U methods, we show that the coupling between a pair of Cr atoms substituted in GaN nanoholes is ferromagnetic. The interaction between... (Read more)
- 231. Phys. Rev. B 75, 075316 (2007) , “Theoretical investigation of a Mn-doped Si/Ge heterostructure”, J. T. Arantes, Antônio J. R. da Silva, A. Fazzio, and A. AntonelliWe investigate, through ab initio density-functional theory calculations, the electronic and structural properties of neutral Mn impurities at tetrahedral interstitial and substitutional sites in both Si and Ge layers of a Si/Ge heterostructure. We conclude that substitutional Mn at the Ge... (Read more)
- 232. Phys. Rev. B 75, 075420 (2007) , “Ab initio study of hydrogen interaction with pure and nitrogen-doped carbon nanotubes”, Zhiyong Zhang and Kyeongjae ChoDetailed studies of mechanisms for hydrogen dissociative adsorption and diffusion on pure and nitrogen-doped (8, 0) carbon nanotubes are carried out using the first-principles density functional theory method. (1) For pure carbon nanotubes, we have identified the energetically most favorable... (Read more)
- 233. Phys. Rev. B 75, 085204 (2007) , “Internal structure of the neutral donor-bound exciton complex in cubic zinc-blende and wurtzite semiconductors”, Bernard Gil, Pierre Bigenwald, Mathieu Leroux, Plamen P. Paskov, and Bo MonemarWe calculate the fine structure splitting of the near band edge donor-bound excitons in major cubic semiconductors using an approach inspired by an earlier one that consists in replacing the Morse potential by a Kratzer one, in order to account for the repulsion between the donor and the hole. A... (Read more)
- 234. Phys. Rev. B 75, 085416 (2007) , “Ab initio theoretical study of hydrogen and its interaction with boron acceptors and nitrogen donors in single-wall silicon carbide nanotubes”, A. GaliSilicon carbide nanotubes have a great potential for biological applications. It is of interest to explore the electronic properties of these nanotubes, and how those are modified in the presence of impurities. Hydrogen is a common impurity that can appear during the growth of silicon carbide... (Read more)
- 235. Phys. Rev. B 75, 085439 (2007) , “Real-space investigation of fast diffusion of hydrogen on Si(001) by a combination of nanosecond laser heating and STM”, C. H. Schwalb, M. Lawrenz, M. Dürr,, and U. HöferThe rearrangement of silicon dangling bonds induced by pulsed laser heating of monohydride-covered Si(001) surfaces has been studied by means of scanning tunneling microscopy (STM). The initial configurations, which were created by laser-induced thermal desorption, consist of isolated pairs of... (Read more)
- 236. Phys. Rev. B 75, 113401 (2007) , “Enhancing the topological structures of defected carbon nanotubes with adsorbed hydrocarbon radicals at low temperatures”, R. L. Zhou, H. Y. He, and B. C. PanRealistic carbon nanotubes (CNTs) contain various structural defects by which the physical and chemical properties of the CNTs are significantly influenced. So, lowering the defect densities is of importance for the potential application of the CNTs. By performing tight-binding molecular dynamics... (Read more)
- 237. Phys. Rev. B 75, 115113 (2007) , “Structural, electronic, and magnetic properties of Mn-doped Ge nanowires by ab initio calculations”, J. T. Arantes, Antônio J. R. da Silva, and A. FazzioUsing ab initio total energy density-functional theory calculations, we investigated the electronic, structural, and magnetic properties of manganese-doped germanium nanowires. The nanowires have been constructed along the [110] direction and the dangling bonds on the surface have been... (Read more)
- 238. Phys. Rev. B 75, 115127 (2007) , “Molecular microelectrostatic view on electronic states near pentacene grain boundaries”, Stijn Verlaak and Paul HeremansGrain boundaries are the most inevitable and pronounced structural defects in pentacene films. To study the effect of those structural defects on the electronic state distribution, the energy levels of a hole on molecules at and near the defect have been calculated using a submolecular... (Read more)
- 239. Phys. Rev. B 75, 115201 (2007) , “Atomistic modeling of the (a+c)-mixed dislocation core in wurtzite GaN”, I. Belabbas, A. Béré, J. Chen, S. Petit, M. Akli Belkhir, P. Ruterana, and G. NouetAn atomistic simulation of the threading (a+c)-mixed dislocation core in wurtzite GaN has been carried out. Starting from models generated in the framework of continuum elasticity theory, two core configurations are obtained independently by using an empirical potential and a... (Read more)
- 240. Phys. Rev. B 75, 115205 (2007) , “Stability of I3 complexes in III-V compound semiconductors by tight-binding molecular dynamics”, G. Zollo and F. GalaIntrinsic interstitials in GaAs are known to have a large formation energy that makes their concentration almost negligible in as-grown materials. However, interstitials must be explicitly considered in implanted GaAs where collision cascades, induced by the energetic ions, produce a large amount of... (Read more)
- 241. Phys. Rev. B 75, 115206 (2007) , “Local-density-functional calculations of the vacancy-oxygen center in Ge”, A. Carvalho, R. Jones, J. Coutinho, V. J. B. Torres, S. Öberg, J. M. Campanera Alsina, M. Shaw, and P. R. BriddonWe carry out a comprehensive density-functional study of the vacancy-oxygen (VO) center in germanium using large H-terminated Ge clusters. The importance of a nonlinear core correction to account for the involvement of the 3d electrons in Ge-O bonds is discussed. We calculate the electrical... (Read more)
- 242. Phys. Rev. B 75, 115418 (2007) , “Early stages of radiation damage in graphite and carbon nanostructures: A first-principles molecular dynamics study”, Oleg V. Yazyev, Ivano Tavernelli, Ursula Rothlisberger, and Lothar HelmUnderstanding radiation-induced defect formation in carbon materials is crucial for nuclear technology and for the manufacturing of nanostructures with desired properties. Using first-principles molecular dynamics, we perform a systematic study of the nonequilibrium processes of radiation damage in... (Read more)
- 243. Phys. Rev. B 75, 134106 (2007) , “Exact linear response of reacting thermal defects driven by creation processes”, C. P. FlynnThe exact, linear response at steady state is calculated for reacting, but otherwise noninteracting, thermal defects driven by defect creation processes. The theory applies to vacancies and interstitials in the bulk, or to adatoms and advacancies on surface terraces. A wide variety of possible... (Read more)
- 244. Phys. Rev. B 75, 144102 (2007) , “Theoretical investigation of nitrogen substitution in cubic zirconia”, Thomas BredowNitrogen substitution of oxygen ions in cubic zirconia was studied theoretically at density functional level. Nitrogen contents of 12.5 and 3.1% were studied with Zr8O16−mN2 and Zr32O64−mN2 supercells. For... (Read more)
- 245. Phys. Rev. B 75, 144103 (2007) , “Multiscale modeling of point defects in Si-Ge(001) quantum wells”, B. Yang and V. K. TewaryA computationally efficient hybrid Green's function (GF) technique is developed for multiscale modeling of point defects in a trilayer lattice system that links seamlessly the length scales from lattice (subnanometers) to continuum (bulk). The model accounts for the discrete structure of the lattice... (Read more)
- 246. Phys. Rev. B 75, 144108 (2007) , “Interaction mechanism between edge dislocations and asymmetrical tilt grain boundaries investigated via quasicontinuum simulations”, T. Shimokawa, T. Kinari, and S. ShintakuThe interactions between edge dislocations and grain boundaries—dislocation pileup, dislocation absorption, and dislocation transmission—are studied by performing quasicontinuum simulations. The 112 asymmetrical tilt grain boundaries with different misorientation angles are used. The... (Read more)
- 247. Phys. Rev. B 75, 144404 (2007) , “Ferromagnetism in Fe-doped ZnO nanocrystals: Experiment and theory”, Debjani Karmakar, S. K. Mandal, R. M. Kadam, P. L. Paulose, A. K. Rajarajan, T. K. Nath, A. K. Das, I. Dasgupta, and G. P. DasFe-doped ZnO nanocrystals are successfully synthesized and structurally characterized by using x-ray diffraction and transmission electron microscopy. Magnetization measurements on the same system reveal a ferromagnetic to paramagnetic transition temperature above 450 K with a low-temperature... (Read more)
- 248. Phys. Rev. B 75, 193203 (2007) , “Hf defects in c-Si and their importance for the HfO2/Si interface: Density-functional calculations”, Wanderlã L. Scopel, Antônio J. R. da Silva, and A. FazzioWe have studied, using ab initio density functional theory calculations, substitutional and interstitial Hf impurities in c-Si, for various charge states. Our results indicate that (1) the tetrahedral interstitial defect is energetically more favorable than the substitutional and (2)... (Read more)
- 249. Phys. Rev. B 75, 193409 (2007) , “Magnetic properties of vacancies in a graphitic boron nitride sheet by first-principles pseudopotential calculations”, M. S. Si and D. S. XueWe use ab initio methods to calculate the magnetic properties of vacancies in a graphitic boron nitride sheet (g-BN). By applying a full spin-polarized description to the system, we demonstrate that the nitrogen vacancy (VN) or the boron vacancy (VB)... (Read more)
- 250. Phys. Rev. B 75, 195206 (2007) , “Effect of defect-enhanced molecular oxygen adsorption on the imbalance of hole versus electron mobility in conjugated polymers”, Chi-Ken Lu, Shu-Ting Pi, and Hsin-Fei MengThe generally observed higher hole mobility relative to electron mobility in conjugated polymers is explained with the defects and adsorbed molecular oxygen. Adsorption of the extrinsic molecular oxygen leads to that electrons are bound more tightly than holes by the traps in the originally... (Read more)
- 251. Phys. Rev. B 75, 195208 (2007) , “Supercell and cluster density functional calculations of the thermal stability of the divacancy in germanium”, C. Janke, R. Jones, S. Öberg, and P. R. BriddonLarge vacancy clusters, or voids, formed during crystal growth have been reported in Ge. The divacancy is a precursor to such clusters, and is believed to be stable up to 150 or 180 °C. It is also believed to form in Ge irradiated at room temperature where single vacancies are mobile. Density... (Read more)
- 252. Phys. Rev. B 75, 195215 (2007) , “Effect of Co and O defects on the magnetism in Co-doped ZnO: Experiment and theory”, G. S. Chang, E. Z. Kurmaev, D. W. Boukhvalov, L. D. Finkelstein, S. Colis, T. M. Pedersen, A. Moewes, and A. DiniaThe electronic structure of Zn1−xCoxO (x=0.02, 0.06, and 0.10) diluted magnetic semiconductors is investigated using soft x-ray emission spectroscopy and first-principles calculations. X-ray absorption and emission measurements reveal that most Co... (Read more)
- 253. Phys. Rev. B 75, 195335 (2007) , “Computational and experimental imaging of Mn defects on GaAs (110) cross-sectional surfaces”, A. Stroppa, X. Duan, M. Peressi, D. Furlanetto, and S. ModestiWe present a combined experimental and computational study of the (110) cross-sectional surface of Mn δ-doped GaAs samples. We focus our study on three different selected Mn defect configurations not previously studied in detail, namely surface interstitial Mn, isolated and in pairs, and... (Read more)
- 254. Phys. Rev. B 75, 245202 (2007) , “Identification of positively charged carbon antisite-vacancy pairs in 4H-SiC”, T. Umeda, J. Ishoya, T. Ohshima, N. Morishita, H. Itoh, and A. GaliAn antisite-vacancy pair and a monovacancy are a set of fundamental stable and/or metastable defects in compound semiconductors. Theory predicted that carbon antisite-vacancy pairs would be much more stable in p-type SiC than silicon vacancies and that they would be a common defect. However,... (Read more)
- 255. J. Appl. Phys. 101, 024101 (2007) , “Defect states in the high-dielectric-constant gate oxide HfSiO4”, K. Xiong, Y. Du, K. Tse, and J. RobertsonHafnium silicate has a high dielectric constant and is a leading candidate to act as a gate dielectric. The defect energy levels have been calculated. The oxygen vacancy is found to give rise to Si-like levels which lie within the band gap of Si. The vacancy states are very localized and are... (Read more)
- 256. Appl. Phys. Lett. 90, 021920 (2007) , “Hydrogen passivation of nitrogen in GaNAs and GaNP alloys: How many H atoms are required for each N atom?”, I. A. Buyanova, W. M. Chen, M. Izadifard, S. J. Pearton, C. Bihler, M. S. Brandt, Y. G. Hong, and C. W. TuSecondary ion mass spectrometry and photoluminescence are employed to evaluate the origin and efficiency of hydrogen passivation of nitrogen in GaNAs and GaNP. The hydrogen profiles are found to closely follow the N distributions, providing unambiguous evidence for their preferential binding as the... (Read more)
- 257. Appl. Phys. Lett. 90, 023112 (2007) , “Electrically tunable defects in metallic single-walled carbon nanotubes”, Ji-Yong ParkA defect whose electron transmission probability can be controlled by electric field is intentionally created on a metallic single-walled carbon nanotube (SWCNT) with a voltage pulse from a tip of an atomic force microscope (AFM). Localized characteristics of the created defect are elucidated with... (Read more)
- 258. Appl. Phys. Lett. 90, 032906 (2007) , “Large scale ab initio molecular dynamics simulations of hydrogen-induced degradation of Ta diffusion barriers in ultralow-k dielectric systems”, Ling Dai, V. B. C. Tan, Shuo-Wang Yang, Ping Wu, and Xian-Tong ChenIn ultralow-k dielectric systems, the porous dielectrics are normally sealed by a SiC film before the deposition of a Ta diffusion barrier layer. However, the Ta barrier effects are negated when the SiC films are fabricated by plasma-enhanced chemical vapor deposition (PECVD). Through large... (Read more)
- 259. Appl. Phys. Lett. 90, 042105 (2007) , “Stress induced leakage current mechanism in thin Hf-silicate layers”, A. Paskaleva, M. Lemberger, and A. J. BauerStress induced leakage current (SILC) in thin Hf-silicate layers and the mechanisms of its creation are examined. A very strong polarity and thickness dependence as well as partial recovery of SILC are observed. It is suggested that the trapping in preexisting sites influences SILC by two ways: (1)... (Read more)
- 260. phys. stat. sol. (b) 245, 1298-1314 (2008) , “EPR identification of intrinsic defects in SiC”, J. Isoya, T. Umeda, N. Mizuochi, N. T. Son, E. Janzen, T. OhshimaThe structure determination of intrinsic defects in 4H-SiC, 6H-SiC, and 3C-SiC by means of EPR is based on measuring the angular dependence of the 29Si/13C hyperfine (HF) satellite lines, from which spin densities, sp-hybrid ratio, and p-orbital direction can be determined over... (Read more)Si SiC diamond| EPR Theory electron-irradiation thermal-meas./anneal-exp.| +1 -1 0(neutral) 1.0eV~ 13C 29Si C1h C3v Carbon Csi D2d EI5/6 HEI1 HEI9/10 P6/7 Silicon T1 Td Tv2a V1/2/3 Vc Vsi antisite dangling-bond mono(=1) motional-effect n-type p-type pair(=2) quartet semi-insulating spin-relaxation triplet vacancy .inp files: SiC/Baranov/Baranov_g.inp SiC/EI5_C1h/5.inp SiC/EI5_C3v/5.inp SiC/EI6_RT/6.inp SiC/HEI10/HEI10a.inp SiC/HEI10/HEI10b.inp SiC/HEI1_C1h/1.inp SiC/HEI9/HEI9a.inp SiC/HEI9/HEI9b.inp SiC/SI5_C1h/4.inp SiC/Ky2/Ky2.inp SiC/Tv2a/Main.INP SiC/Vsi-_II_4H/Main.INP SiC/Vsi-_II_6H/Main.INP SiC/Vsi-_I_4H/Main.INP SiC/Vsi-_I_6H/Main.INP | last update: Takahide Umeda
- 261. J.Am.Chem.Soc. 130, 48 (2008) , ACS , “Enhanced Ferromagnetism and Tunable Saturation Magnetization of Mn/C Codoped GaN Nanostructures Synthesized by Carbothermal Nitridation”, Zeyan Wang, Baibiao Huang, Lin Yu, Ying Dai, Peng Wang, Xiaoyan Qin, Xiaoyang Zhang, Jiyong Wei, Jie Zhan, Xiangyang Jing, Haixia Liu, and Myung-Hwan WhangboMn/C-codoped GaN nanostructures were synthesized by carbothermal nitridation with active charcoal as the carbon source. Nanostructures such as zigzag nanowires and nanoscrews were observed by varying the reaction time and the C/Ga molar ratio of the starting material used for the synthesis. The structures and morphologies of the as-grown samples were characterized by X-ray diffraction, scanning electron microscopy, and high-resolution transmission electron microscopy measurements. The doping of both Mn and C in the GaN matrix was confirmed by X-ray photoelectron spectroscopy measurements, and the ferromagnetic properties of Mn/C-codoped GaN samples were confirmed by room-temperature magnetization measurements. The saturation magnetization of Mn/C-codoped GaN increases steadily with increasing C/Ga molar ratio of the starting material at a rate of ~0.023 emu/g per C/Ga molar ratio, and the ferromagnetism of Mn/C-codoped GaN can be stronger than that of Mn-doped GaN by a factor of ~40. A plausible growth mechanism was proposed, and the role of carbon codoping in tuning the morphology and ferromagnetic property was discussed. Our work suggests that carbon doping in the GaN matrix favors the N sites over the Ga sites, Mn/C-codoping in the GaN matrix is energetically favorable, and the C-codoping strongly enhances the preference of the FM coupling to the AFM coupling between the two doped Mn sites. These suggestions were probed on the basis of first-principles density functional theory electronic structure calculations for a number of model doped structures constructed with a 32-atom 2 × 2 × 2 supercell. (Read more)
« Previous
Next »
(261 hits, 1/1)
Showing
10, 25, 50, 100, 500, 1000, all papers per page.
Sort by:
last publication date,
older publication date,
last update date.
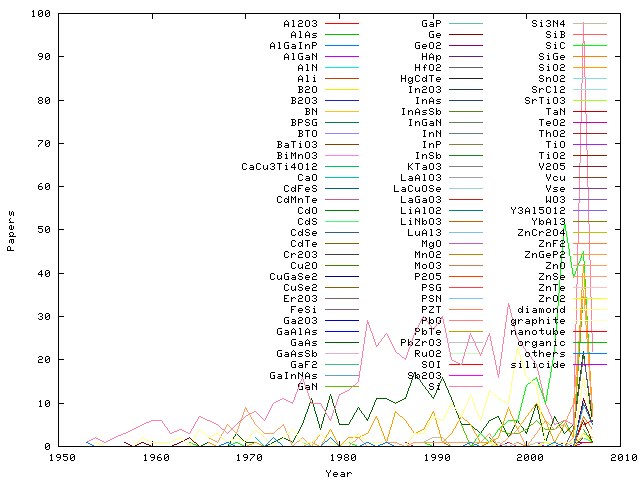
Updated at 2010-07-20 16:50:39
(view as: tree
,
cloud
)
| 1329 | untagged |
Materials
(111 tags)
Others(101 tags)
Technique
(46 tags)
Details
(591 tags)
Bond(35 tags)
Defect(interstitial)(18 tags)
Defect(vacancy)(15 tags)
Defect-type(19 tags)
Element(65 tags)
Energy(8 tags)
Isotope(56 tags)
Label(303 tags)
Sample(17 tags)
Spin(8 tags)
Symmetry(15 tags)